El acrónimo SMA en el campo de la neurología puede referirse a dos condiciones fundamentalmente distintas, cada una con implicaciones y manifestaciones muy diferentes en el sistema nervioso. Es crucial entender esta dualidad para evitar confusiones y comprender correctamente las investigaciones, diagnósticos y tratamientos asociados. Por un lado, encontramos la Atrofia Muscular Espinal, una enfermedad genética que afecta las neuronas motoras de la médula espinal. Por otro lado, existe el Síndrome del Área Motora Suplementaria, un fenómeno neurológico que surge tras lesiones o intervenciones en una región específica del cerebro. Aunque comparten el mismo acrónimo, sus causas, síntomas y pronósticos son radicalmente diferentes.
Atrofia Muscular Espinal (AME)
La Atrofia Muscular Espinal (AME), conocida como SMA por sus siglas en inglés (Spinal Muscular Atrophy), es un trastorno genético hereditario que impacta directamente a las neuronas motoras. Estas células nerviosas, localizadas principalmente en la médula espinal, son responsables de controlar el movimiento muscular voluntario. En la AME, estas neuronas se degeneran o no funcionan correctamente, lo que impide que envíen las señales necesarias a los músculos.
Dado que los músculos no reciben las señales nerviosas adecuadas, no pueden contraerse ni realizar movimientos voluntarios. Esta inactividad lleva a que los músculos se debiliten y se encojan con el tiempo, un proceso conocido como atrofia. La debilidad muscular tiende a ser más pronunciada en los músculos cercanos al centro del cuerpo (músculos proximales), como los de los hombros, caderas, muslos y parte superior de la espalda, aunque también puede afectar a los músculos más distantes (distales) en algunas formas. Es importante destacar que la AME no afecta las neuronas sensoriales ni, en la mayoría de los casos, el intelecto. De hecho, se ha observado que muchas personas con AME poseen una inteligencia notablemente alta.
La AME es una enfermedad relativamente común dentro de los trastornos genéticos graves de la infancia, afectando aproximadamente a 1 de cada 6,000 a 10,000 recién nacidos. Es una causa significativa de mortalidad infantil, aunque los avances recientes en las terapias están mejorando el pronóstico para muchos niños.
Síntomas de la Atrofia Muscular Espinal
Los síntomas de la AME varían considerablemente en severidad, desde leves hasta gravemente incapacitantes, pero siempre implican debilidad en los músculos que controlan el movimiento voluntario. Los músculos involuntarios, como los del corazón, los vasos sanguíneos y el tracto digestivo, generalmente no se ven afectados.
- Debilidad muscular progresiva, más notoria en hombros, caderas, muslos y espalda.
- Dificultad para realizar hitos motores como levantar la cabeza, sentarse, gatear o caminar, dependiendo del tipo y la edad de inicio.
- Temblores finos en los dedos (fasciculaciones).
- Problemas respiratorios debido a la debilidad de los músculos intercostales y del diafragma.
- Dificultad para tragar y alimentarse adecuadamente.
- Desarrollo de escoliosis (curvatura de la columna vertebral) debido a la debilidad de los músculos de la espalda.
La progresión de la enfermedad puede afectar gravemente la función respiratoria y de deglución, lo que representa un riesgo vital significativo, especialmente en los tipos de inicio temprano.
Causas y Factores de Riesgo
La AME es un trastorno genético hereditario. La mayoría de las formas de AME son causadas por mutaciones en el gen de la neurona motora de supervivencia 1 (SMN1), localizado en el cromosoma 5. Esta mutación resulta en una producción insuficiente de la proteína SMN, que es esencial para la supervivencia y el funcionamiento normal de las neuronas motoras. Sin suficiente proteína SMN, las neuronas motoras se degeneran, interrumpiendo la comunicación entre los nervios y los músculos.
Existen formas menos comunes de AME que no están vinculadas a mutaciones en el gen SMN1 o deficiencia de la proteína SMN. Estas formas pueden variar en severidad y a veces afectan músculos más distantes del centro del cuerpo.
Tipos de Atrofia Muscular Espinal
La AME se clasifica tradicionalmente en tipos (0, 1, 2, 3, 4) basándose en la edad de inicio de los síntomas y la máxima capacidad motora alcanzada. Generalmente, cuanto más tarde aparecen los síntomas, menor es la severidad y mejor el pronóstico para la función motora.
| Tipo de AME | Edad de Inicio | Severidad | Capacidades Motoras Típicas | Pronóstico Vital (sin tratamiento) |
|---|---|---|---|---|
| Tipo 0 (Congénita) | Antes del nacimiento | Muy Severa | Movimientos fetales disminuidos. Debilidad severa al nacer. | Muerte al nacer o en el primer mes. |
| Tipo 1 (Infantil Severa / Werdnig-Hoffman) | 0-6 meses | Severa | No alcanzan la capacidad de sentarse sin apoyo. Dificultad para tragar y respirar. | Muerte antes de los 2 años. |
| Tipo 2 (Intermedia / Dubowitz) | 6-18 meses | Intermedia | Pueden sentarse sin apoyo pero no caminar. Debilidad más pronunciada en las piernas. | Supervivencia hasta la adolescencia tardía o edad adulta joven (20s-30s). |
| Tipo 3 (Juvenil / Kugelberg-Welander) | Después de los 18 meses (puede ser en la adolescencia) | Leve a Moderada | Pueden caminar, pero pueden perder esta capacidad con el tiempo. Debilidad en miembros inferiores. | Generalmente no afecta la esperanza de vida. |
| Tipo 4 (Adulta) | Después de los 21 años | Leve | Debilidad muscular de progresión lenta. La mayoría permanece móvil. | No afecta la esperanza de vida. |
Es importante mencionar que, con los tratamientos modernos que modifican la enfermedad, las trayectorias de los pacientes pueden ser significativamente diferentes a las descritas en esta clasificación tradicional.
Diagnóstico de la AME
El diagnóstico de la AME se basa en la evaluación clínica de los síntomas, el historial familiar y pruebas específicas para confirmar la causa genética.
- Prueba Genética: Es el método diagnóstico más común y definitivo. Un análisis de sangre puede detectar las mutaciones en el gen SMN1 responsables de la mayoría de los casos de AME.
- Electromiografía (EMG): Mide la actividad eléctrica de los músculos en respuesta a la estimulación nerviosa. En la AME, muestra patrones característicos de denervación (pérdida de conexión nerviosa).
- Biopsia Muscular: Aunque menos común hoy en día debido a la fiabilidad de las pruebas genéticas, una pequeña muestra de músculo puede ser examinada al microscopio para buscar signos de atrofia y otros cambios característicos.
Tratamiento y Manejo de la AME
Actualmente, existen tratamientos que modifican la enfermedad y han revolucionado el manejo de la AME, mejorando significativamente los resultados para muchos pacientes. Sin embargo, todavía no hay una cura en el sentido tradicional.
- Medicamentos: Se han aprobado varias terapias (como Nusinersen, Onasemnogene abeparvovec y Risdiplam) que actúan sobre el gen SMN, aumentando los niveles de la proteína SMN o reemplazando el gen defectuoso. Estos tratamientos pueden ralentizar o detener la progresión de la enfermedad y, en algunos casos, mejorar la función motora.
- Terapia Física, Ocupacional y de Rehabilitación: Es fundamental para mantener la flexibilidad de las articulaciones, prevenir contracturas, mejorar la circulación y maximizar la función muscular residual. Terapeutas especializados pueden ayudar con ejercicios, estiramientos y estrategias para la vida diaria.
- Dispositivos de Apoyo: Ortesis, corsés, soportes y sillas de ruedas son esenciales para ayudar a los pacientes a mantener la independencia, apoyar la postura (especialmente para la escoliosis) y facilitar la movilidad.
- Asistencia Respiratoria: Muchos pacientes, especialmente en los tipos más severos, requieren soporte para la respiración. Esto puede variar desde ventilación no invasiva durante la noche hasta asistencia ventilatoria continua.
- Manejo Nutricional: Las dificultades para tragar pueden requerir adaptaciones en la dieta, técnicas de alimentación especializadas o el uso de sondas de alimentación para asegurar una nutrición adecuada y prevenir la aspiración (paso de alimentos o líquidos a las vías respiratorias).
El manejo de la AME requiere un enfoque multidisciplinario que involucre neurólogos, neumólogos, gastroenterólogos, fisioterapeutas, terapeutas ocupacionales, genetistas y otros especialistas.
Preguntas Frecuentes sobre la Atrofia Muscular Espinal
Aquí respondemos algunas preguntas comunes sobre la AME:
¿La AME es contagiosa?
No, la AME es un trastorno genético y no se puede contagiar de una persona a otra.
¿La AME afecta la inteligencia?
Generalmente no. La AME afecta las neuronas motoras, no las neuronas cerebrales responsables del intelecto. De hecho, muchas personas con AME son intelectualmente muy capaces.
¿Hay cura para la AME?
Actualmente no existe una cura que revierta completamente el daño causado por la enfermedad. Sin embargo, las terapias disponibles modifican la enfermedad y pueden detener o ralentizar su progresión, mejorando significativamente la calidad y esperanza de vida, especialmente si se inician tempranamente.
¿Cómo se hereda la AME?
La forma más común de AME se hereda de forma autosómica recesiva. Esto significa que una persona debe heredar una copia mutada del gen SMN1 de cada uno de sus padres para desarrollar la enfermedad. Los padres que portan una sola copia mutada generalmente no presentan síntomas (son portadores).
Síndrome del Área Motora Suplementaria (SMA)
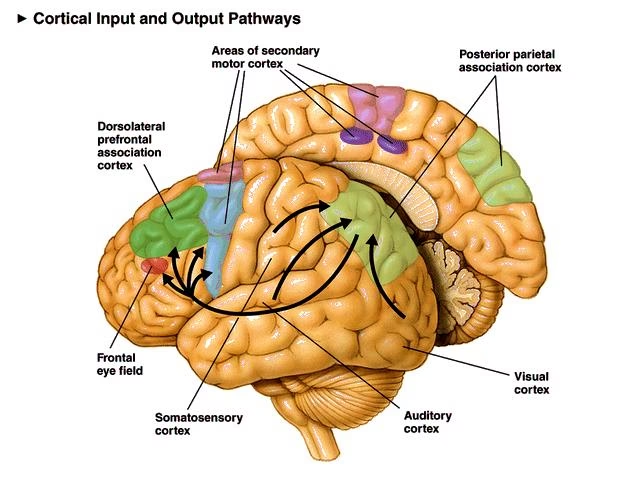
Por otro lado, el acrónimo SMA también se utiliza en neurología para referirse al Área Motora Suplementaria del cerebro (Supplementary Motor Area). Esta región, localizada en la parte posterior del giro frontal superior (aproximadamente el área de Brodmann 6), juega un papel crucial en la planificación, iniciación y ejecución de movimientos complejos, especialmente aquellos que son autoiniciados o que involucran secuencias y coordinación bimanual.
El Síndrome del Área Motora Suplementaria es una condición neurológica que puede ocurrir tras la resección quirúrgica o la lesión de esta área cerebral. Es un fenómeno particularmente intrigante en neurocirugía debido a su naturaleza típicamente transitoria.
Características del Síndrome del Área Motora Suplementaria
El síndrome clásico, observado después de la resección unilateral del SMA, se caracteriza por una serie de síntomas que, aunque pueden ser alarmantes inicialmente, tienden a resolverse con el tiempo.
- Akinesia Global: Una marcada dificultad o incapacidad para iniciar movimientos voluntarios, predominantemente en el lado del cuerpo contralateral a la lesión cerebral.
- Mutismo: Reducción o ausencia de habla espontánea, aunque la comprensión del lenguaje suele estar preservada.
- Fuerza Muscular Preservada: A diferencia de la AME, la fuerza muscular básica no está afectada; el problema radica en la capacidad de iniciar y coordinar el movimiento.
- Normo o Hiporreflexia: Reflejos normales o disminuidos.
La característica más notable de este síndrome es que estos síntomas, a menudo severos en las primeras semanas después de la lesión o cirugía, tienden a resolverse casi por completo en un período de semanas a meses. El déficit residual más común y persistente es una dificultad en los movimientos bimanuales alternos, es decir, la capacidad de realizar movimientos coordinados y alternados con ambas manos de forma fluida.
El Área Motora Suplementaria y su Función
El SMA es un área de asociación motora con ricas conexiones con otras regiones corticales (corteza motora primaria, premotora, cíngulo) y subcorticales (ganglios basales, tálamo, cerebelo). Tiene una organización somatotópica, con representaciones del cuerpo dispuestas de adelante hacia atrás (cara, miembros superiores, miembros inferiores).
Se cree que el SMA está involucrado en la planificación motora de alto nivel, la preparación para el movimiento, la secuenciación de acciones y, crucialmente, en la coordinación de movimientos complejos que involucran ambos lados del cuerpo. También se ha sugerido su papel en la iniciación del movimiento autogenerado (sin una señal externa) y en la supresión de movimientos no deseados.
Causa y Mecanismos de Recuperación
El síndrome ocurre típicamente después de la resección quirúrgica de tumores localizados en el SMA o tras lesiones isquémicas (infartos) que afectan esta área. La severidad y la duración del síndrome pueden estar relacionadas con el tamaño de la resección y la proximidad a otras áreas motoras críticas, así como a las vías de sustancia blanca que conectan el SMA con otras regiones.
La recuperación de la akinesia y el mutismo es un ejemplo notable de plasticidad cerebral. Se cree que otras áreas cerebrales, particularmente el SMA contralateral (en el hemisferio no afectado) y la corteza premotora lateral, asumen funciones compensatorias a lo largo del tiempo. La fuerte conexión entre los dos SMA a través del cuerpo calloso parece ser fundamental para esta recuperación, aunque la persistencia del déficit en los movimientos bimanuales alternos sugiere que la función bilateral del SMA es necesaria para tareas motoras complejas que requieren una coordinación precisa entre ambos lados.
Comparación con Otras Condiciones
La observación del Síndrome del Área Motora Suplementaria ha proporcionado información valiosa sobre la función del SMA, a menudo comparándola con déficits observados en otras enfermedades neurológicas:
- Enfermedad de Parkinson (EP): Los pacientes con EP a menudo presentan akinesia (dificultad para iniciar movimientos) y problemas con los movimientos alternos rápidos. Se ha observado una disminución de la actividad en el SMA en pacientes con EP. La comparación sugiere que una disfunción en el circuito de los ganglios basales que se proyecta al SMA contribuye a los síntomas motores de la EP.
- Trastornos de Tics (como el Síndrome de Tourette): Los tics son movimientos o vocalizaciones involuntarias. En contraste con la hipoactividad del SMA en la EP, se ha observado una actividad aumentada o alterada en el SMA en pacientes con tics. Esto ha llevado a la hipótesis de que el SMA está involucrado tanto en la iniciación del movimiento como en su supresión, y un desequilibrio en esta función podría contribuir a la aparición de tics.
Estas comparaciones refuerzan la idea de que el SMA juega un papel complejo en el control motor, participando en la preparación, iniciación, secuenciación y coordinación de movimientos, y manteniendo un equilibrio entre la facilitación y la inhibición de la actividad motora.
Preguntas Frecuentes sobre el Síndrome del Área Motora Suplementaria
Aquí respondemos algunas preguntas comunes sobre el Síndrome del SMA:
¿El Síndrome del SMA es permanente?
Los síntomas más severos, como la akinesia global y el mutismo, suelen ser temporales y se resuelven en semanas o meses debido a la plasticidad cerebral. Sin embargo, una dificultad sutil pero persistente en los movimientos bimanuales alternos puede permanecer.
¿Qué causa el Síndrome del SMA?
Generalmente es causado por la resección quirúrgica de tumores localizados en o cerca del Área Motora Suplementaria del cerebro, o por lesiones como infartos en esta región.
¿Afecta la fuerza muscular?
No directamente. La fuerza muscular básica suele estar preservada. El síndrome afecta la capacidad de iniciar y coordinar los movimientos voluntarios.
En conclusión, el término SMA en neurología abarca dos entidades clínicas muy diferentes: la Atrofia Muscular Espinal, una enfermedad genética neuromuscular devastadora, y el Síndrome del Área Motora Suplementaria, un fenómeno cerebral transitorio relacionado con lesiones en una región específica del cerebro. Comprender esta distinción es fundamental para navegar el complejo campo de la neurología y sus diversas patologías.
Si quieres conocer otros artículos parecidos a SMA en Neurología: Dos Significados Clave puedes visitar la categoría Neurología.