El término amiloide se refiere a depósitos proteicos que se pliegan de manera anómala y se agregan para formar estructuras fibrosas altamente organizadas. Aunque inicialmente se pensó que solo aparecían en contextos patológicos, hoy sabemos que algunas formaciones amiloides cumplen funciones biológicas normales. Sin embargo, su notoriedad proviene principalmente de su asociación con una amplia gama de enfermedades, muchas de ellas graves y debilitantes, incluyendo trastornos neurodegenerativos.
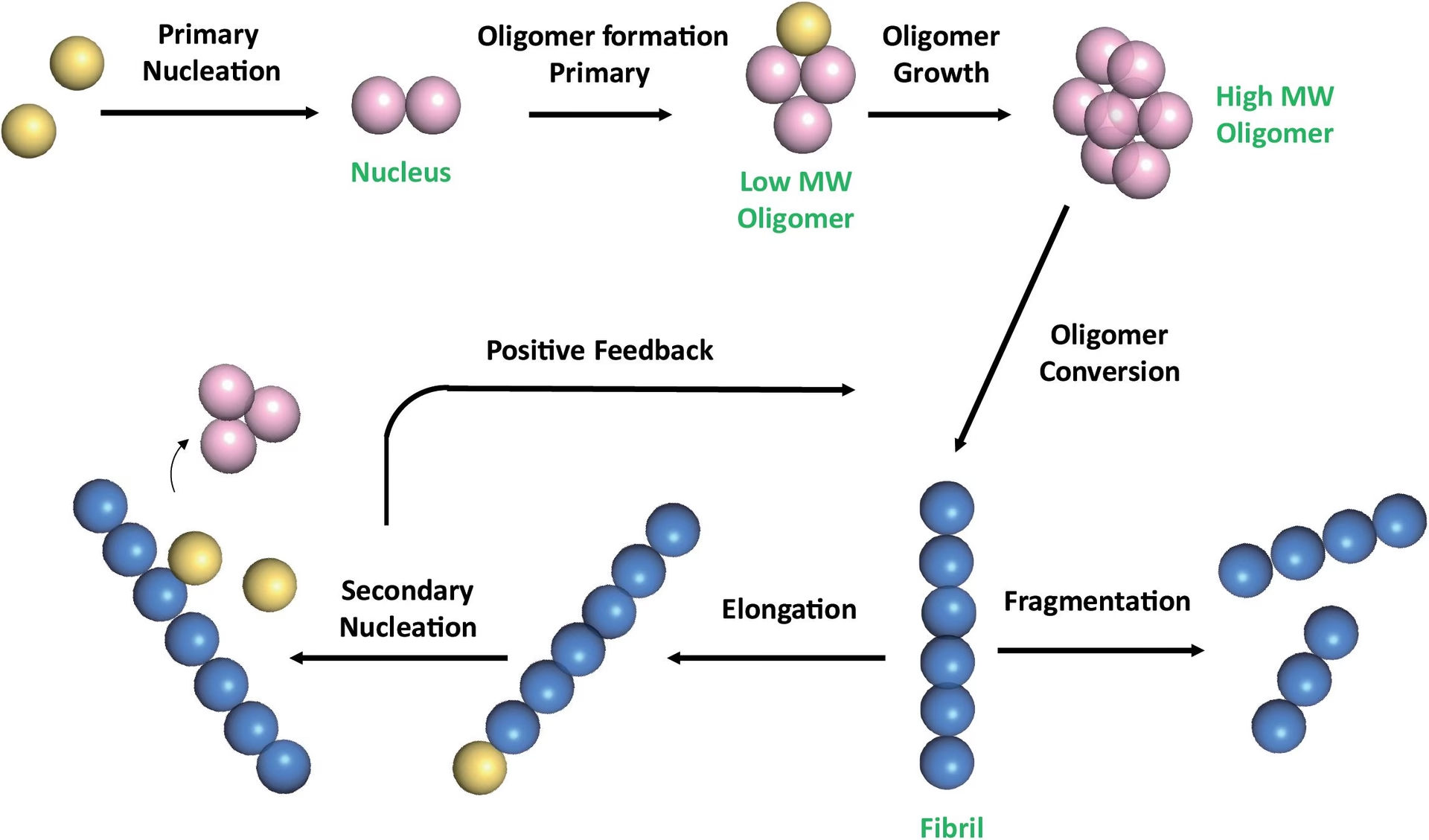
Estas estructuras, compuestas por cientos o miles de péptidos o proteínas individuales (monómeros) que se polimerizan en largas fibras, representan un desafío significativo en la investigación biomédica. Comprender su naturaleza química, los intrincados mecanismos por los que se forman y, crucialmente, cómo ejercen su toxicidad, es fundamental para desarrollar estrategias terapéuticas efectivas.
- ¿Qué es Exactamente el Amiloide?
- Proteínas Capaces de Formar Amiloide
- El Intrincado Proceso de Formación de Fibrillas Amiloides
- La Influencia Crucial de la Secuencia de Aminoácidos
- El Enigma de la Toxicidad del Amiloide
- Detección y Diagnóstico: Tinción Histológica del Amiloide
- Tabla Comparativa de Fases de Formación de Amiloide
- Preguntas Frecuentes sobre el Amiloide
¿Qué es Exactamente el Amiloide?
Desde un punto de vista químico, el amiloide no es una sustancia única, sino un estado físico en el que pueden encontrarse diversas proteínas o péptidos. Lo que define al amiloide es su estructura característica: un plegamiento anómalo que adopta predominantemente una conformación de lámina beta cruzada. Esta estructura permite que las moléculas individuales se apilen y unan de forma muy estable, dando lugar a las fibrillas amiloides, que son muy resistentes a la degradación.
Aunque la secuencia de aminoácidos de las proteínas que forman amiloide puede ser muy diversa, la estructura final de la fibra amiloide es notablemente similar entre diferentes tipos de amiloidosis. Es esta estructura compartida la que permite su detección mediante tinciones específicas y explica algunas de sus propiedades físicas y biológicas.
Proteínas Capaces de Formar Amiloide
Una variedad sorprendente de proteínas y péptidos en el cuerpo humano pueden, bajo ciertas condiciones, adoptar el plegamiento amiloide. Algunas son bien conocidas por su implicación en enfermedades:
- El péptido Beta-Amiloide (Aβ), asociado a la enfermedad de Alzheimer.
- La proteína Tau, que forma ovillos neurofibrilares también en Alzheimer y otras taupatías.
- La Alfa-sinucleína, implicada en la enfermedad de Parkinson y la demencia con cuerpos de Lewy.
- La proteína Priónica (PrP), responsable de las enfermedades priónicas como la de Creutzfeldt-Jakob.
- La Amilina (o IAPP), un péptido asociado a la diabetes tipo 2.
- La Huntingtina, una proteína con repeticiones de glutamina expandidas, causante de la enfermedad de Huntington.
La capacidad de una proteína para formar amiloide depende en gran medida de su secuencia de aminoácidos y de las condiciones del entorno. Pequeñas variaciones en la secuencia pueden determinar si una proteína es amiloidogénica o no, como se observa al comparar la amilina humana (amiloidogénica) con la de rata o ratón (no amiloidogénica debido a sustituciones de prolina).
El Intrincado Proceso de Formación de Fibrillas Amiloides
La formación de las fibrillas amiloides es un proceso complejo que generalmente sigue una cinética sigmoidal cuando se representa la cantidad de fibrillas frente al tiempo. Esto refleja tres fases distintas:
- Fase de Latencia (o Nucleación): Es la fase inicial, aparentemente lenta. Durante este período, las proteínas monoméricas (plegadas o desplegadas parcialmente) deben superar una barrera energética para formar un 'núcleo' o 'semilla'. Este paso es termodinámicamente desfavorable y es el cuello de botella del proceso. La formación de este núcleo, ya sea a partir de monómeros (nucleación primaria) o a través de oligómeros desorganizados que se reestructuran, es esencial para que la agregación progrese.
- Fase Exponencial (o Crecimiento): Una vez que se han formado suficientes núcleos, las proteínas monoméricas (o a veces oligómeros) se añaden rápidamente a los extremos de estos núcleos, provocando un crecimiento rápido de las fibrillas. La cantidad de fibrillas aumenta drásticamente durante esta fase.
- Fase de Meseta (o Saturación): La velocidad de formación de fibrillas disminuye a medida que la concentración de monómeros libres disponibles para la elongación se agota, o se alcanza un equilibrio entre la formación y la disociación/degradación.
Modelos más recientes y sofisticados de formación de amiloide incorporan eventos secundarios que tienen un impacto significativo en la cinética del proceso, especialmente en la fase exponencial. Estos eventos incluyen la fragmentación de las fibrillas existentes (creando nuevos extremos para la elongación) y la nucleación secundaria, donde la superficie de las fibrillas existentes actúa como catalizador para la formación de nuevos núcleos a partir de monómeros. Estos procesos secundarios crean un mecanismo de retroalimentación positiva que acelera enormemente la formación de fibrillas y son cruciales para entender cómo las agregaciones pueden propagarse.
El estudio de estas fases y eventos, a menudo mediante enfoques cinéticos y modelado matemático, permite determinar las tasas de los diferentes pasos (nucleación primaria, elongación, fragmentación, nucleación secundaria) y comprender cómo diversos factores, como mutaciones, chaperonas o posibles fármacos, influyen en el proceso.
La Influencia Crucial de la Secuencia de Aminoácidos
La capacidad de una proteína para formar amiloide es altamente sensible a su secuencia de aminoácidos. Pequeños cambios, como la sustitución de un solo aminoácido, pueden convertir una proteína no amiloidogénica en una que sí lo es, o viceversa. Esto subraya que, aunque la estructura final de la fibra amiloide es genérica (lámina beta cruzada), el camino para llegar a ella y la propensión a hacerlo están codificados en la secuencia primaria de la proteína.
Regiones ricas en ciertos aminoácidos son particularmente propensas a formar amiloide. Por ejemplo, los polipéptidos ricos en glutamina, como la huntingtina, tienden a agregarse, y la longitud de la cadena de glutaminas se correlaciona inversamente con la edad de inicio en la enfermedad de Huntington. De manera similar, las regiones ricas en residuos hidrofóbicos o con alta propensión a formar estructuras de lámina beta son comunes en muchas proteínas amiloidogénicas, siendo los aminoácidos aromáticos (como fenilalanina, tirosina, triptófano) particularmente favorecedores de la formación de amiloide.
Un fenómeno interesante es la "polimerización cruzada", donde las fibrillas formadas por una proteína pueden inducir o acelerar la agregación de otra proteína diferente. Este proceso, observado in vitro y postulado in vivo, podría explicar la propagación de priones entre especies o la posible conexión estadística entre enfermedades como el Alzheimer y la diabetes tipo 2 (implicando Aβ y amilina).
El Enigma de la Toxicidad del Amiloide
A pesar de décadas de investigación, las razones exactas por las que los depósitos amiloides causan enfermedad no están completamente claras. Inicialmente, se pensó que la simple acumulación física de las fibrillas y la disrupción de la arquitectura tisular eran las causas principales. Sin embargo, un consenso emergente apunta cada vez más a los intermediarios prefibrilares (oligómeros) como las especies más tóxicas, especialmente en enfermedades neurodegenerativas.
No obstante, las fibrillas maduras tampoco son inocuas. Pueden interactuar con las redes de homeostasis proteica celular, servir como reservorios que liberan oligómeros tóxicos, catalizar la formación de nuevos oligómeros tóxicos a través de la nucleación secundaria, y propagarse de una célula a otra o de una región a otra del tejido.
Se han propuesto múltiples mecanismos de toxicidad, y es probable que varios de ellos contribuyan simultáneamente:
- Disregulación del Calcio: Los oligómeros amiloides pueden insertar en las membranas celulares y formar canales iónicos o interactuar con receptores (como NMDA o AMPA), alterando la homeostasis del calcio intracelular.
- Disfunción Mitocondrial: La agregación amiloide se asocia a menudo con la disfunción de las mitocondrias, las centrales energéticas de la célula, lo que lleva a una producción excesiva de especies reactivas de oxígeno (ROS) y puede desencadenar la apoptosis (muerte celular programada).
- Interacción con Componentes Celulares: Los agregados mal plegados pueden interactuar de forma aberrante con una multitud de componentes celulares, incluyendo membranas, receptores de superficie, proteínas solubles, ARN y pequeños metabolitos, interrumpiendo numerosas vías celulares vitales.
- Secuestro de Proteínas Esenciales: Los agregados pueden "secuestrar" otras proteínas esenciales para el funcionamiento celular, incorporándolas a los agregados o impidiendo su interacción con sus parejas funcionales normales.
Dada la diversidad de agregados que se forman (monómeros, oligómeros de diferentes tamaños y conformaciones, fibrillas) y la multitud de interacciones aberrantes que pueden establecer, es muy probable que la toxicidad del amiloide sea un fenómeno multifacético sin un único mecanismo causal universal.
Detección y Diagnóstico: Tinción Histológica del Amiloide
En el ámbito clínico y de investigación, la presencia de depósitos amiloides se detecta típicamente mediante el uso de tinciones especiales que se unen específicamente a la estructura de lámina beta cruzada. Las más comunes son:
- Tioflavina T: Un colorante fluorescente cuya emisión aumenta significativamente al unirse a las fibrillas amiloides.
- Rojo Congo: Este colorante es el "estándar de oro" para el diagnóstico de amiloidosis en tejidos. Al unirse al amiloide, produce una birrefringencia verde manzana característica cuando se observa bajo luz polarizada cruzada.
- NIAD-4: Otro colorante fluorescente utilizado para la detección de amiloide.
Estos colorantes se intercala entre las hebras beta de las fibrillas, cambiando su entorno y propiedades espectroscópicas. Aunque la inmunohistoquímica (usando anticuerpos contra la proteína específica formadora de amiloide) también se utiliza, puede ser desafiante porque la conformación amiloide puede ocultar los epítopos a los que se unen los anticuerpos.
Tabla Comparativa de Fases de Formación de Amiloide
| Fase de Formación del Amiloide | Descripción Clave | Eventos Principales Incluidos (Modelo Moderno) |
|---|---|---|
| Fase de Latencia (Lag) | Período inicial lento. Formación de núcleos (nucleación primaria). | Nucleación Primaria, Inicio de Elongación y Eventos Secundarios. |
| Fase Exponencial (Crecimiento) | Aumento rápido de la cantidad de fibrillas. | Elongación Rápida, Fragmentación y Nucleación Secundaria (a menudo dominantes). |
| Fase de Meseta (Saturación) | La formación de fibrillas se ralentiza o detiene. Monómeros agotados o equilibrio. | Tasa de formación iguala tasa de disociación/degradación. |
Preguntas Frecuentes sobre el Amiloide
- ¿Todas las proteínas pueden formar amiloide?
No, solo aquellas con secuencias o regiones específicas que, bajo ciertas condiciones, tienen la propensión a adoptar el plegamiento de lámina beta cruzada y agregarse. - ¿El amiloide siempre causa enfermedad?
Si bien la mayoría de las veces se asocia a patologías, se ha descubierto que algunas proteínas forman estructuras amiloides funcionales en organismos sanos, cumpliendo roles biológicos específicos. Sin embargo, la acumulación descontrolada sí suele ser perjudicial. - ¿Qué enfermedades están más comúnmente asociadas con el amiloide?
Enfermedades neurodegenerativas como el Alzheimer (péptido Aβ y Tau), Parkinson (alfa-sinucleína) y Huntington (huntingtina), así como enfermedades sistémicas como la Amiloidosis AA, AL, y la diabetes tipo 2 (amilina). - ¿Cómo se detecta el amiloide en un paciente?
El método diagnóstico estándar en tejido es la tinción con Rojo Congo seguida de observación bajo luz polarizada, que muestra la birrefringencia verde manzana. También se usan otras tinciones y, en investigación, técnicas de imagen molecular avanzadas.
En conclusión, el amiloide es un estado de agregación proteica definido por una estructura particular de lámina beta cruzada. Su formación es un proceso cinéticamente complejo que involucra múltiples pasos, desde la nucleación inicial hasta la elongación y eventos secundarios como la fragmentación. Aunque la secuencia de la proteína influye en su propensión a agregarse, la estructura final de la fibra es genérica. La toxicidad asociada al amiloide es multifacética, involucrando probablemente tanto a los oligómeros prefibrilares como a las fibrillas maduras, a través de diversas interacciones celulares. La detección histológica, particularmente con Rojo Congo, sigue siendo fundamental para su diagnóstico. La investigación continua sobre la naturaleza, formación y toxicidad del amiloide es vital para abordar las devastadoras enfermedades que provoca.
Si quieres conocer otros artículos parecidos a Amiloide: Naturaleza, Formación y Toxicidad puedes visitar la categoría Neurociencia.