La depresión, o trastorno depresivo mayor, es una condición que afecta a una proporción significativa de la población, con casi una de cada cinco personas experimentando un episodio depresivo mayor en algún momento de su vida. Lejos de ser simplemente un estado de ánimo bajo, la depresión es un trastorno complejo con profundas raíces en la biología cerebral. La investigación sugiere que no hay una única causa, sino que surge de la interacción acumulativa de factores genéticos heredados, adversidades psicosociales experimentadas en la infancia y el estrés actual o reciente.
Un episodio depresivo mayor se caracteriza por un estado de ánimo persistentemente bajo o una incapacidad para experimentar placer (anhedonia), o ambos, durante más de dos semanas, acompañado de una serie de síntomas cognitivos y vegetativos que causan malestar o deterioro significativo. Aunque el diagnóstico puede hacerse con un solo episodio, la mayoría de las personas afectadas experimentan múltiples episodios a lo largo de su vida. Es importante destacar que la depresión a menudo coexiste con otras enfermedades médicas crónicas como la diabetes, enfermedades cardíacas y trastornos autoinmunes, existiendo una relación bidireccional donde una condición puede influir en el pronóstico de la otra.
- La Hipótesis Monoaminérgica: Un Punto de Partida
- Genes y Vulnerabilidad: El Papel de la Genética
- Factores de Crecimiento Neuronal: BDNF y la Plasticidad Cerebral
- Adversidad Temprana y el Eje HPA: Cómo el Estrés Moldea el Cerebro
- Otros Neurotransmisores Clave: Dopamina y Glutamato
- Cambios Estructurales y Funcionales en el Cerebro Deprimido
- Estrategias de Tratamiento: Abordando la Base Neurológica
- Preguntas Frecuentes
- Conclusión: Hacia un Enfoque Multifacético
La Hipótesis Monoaminérgica: Un Punto de Partida
Tradicionalmente, la investigación sobre la neurobiología del trastorno depresivo mayor se ha centrado en los neurotransmisores monoaminérgicos, principalmente la serotonina y la norepinefrina. La hipótesis monoaminérgica inicial postulaba que las personas deprimidas probablemente tenían niveles bajos de estos neurotransmisores, basándose en que varios antidepresivos aumentan agudamente sus niveles. Sin embargo, a pesar de ser tratamientos de primera línea, los antidepresivos monoaminérgicos no ejercen su beneficio clínico de inmediato y, para algunas personas, no proporcionan ningún alivio.
Estudios más recientes han llevado a una revisión de esta hipótesis. La investigación en fisiopatología a menudo se realiza en personas actualmente deprimidas, lo que dificulta distinguir causa y efecto o mecanismos centrales de epifenómenos. Por ejemplo, encontrar una tasa baja de síntesis de serotonina en pacientes deprimidos podría significar que la baja serotonina causa depresión, que la depresión reduce la síntesis de serotonina, o que un tercer factor es responsable de ambos.
Los estudios experimentales con pacientes en remisión han ayudado a aclarar el papel de la serotonina. La depleción de triptófano (precursor de la serotonina) puede disminuir temporalmente los niveles de serotonina cerebral. En la mayoría de estos estudios, los pacientes en remisión experimentan una breve recaída, especialmente si están tomando medicación o si su remisión es reciente. Esto sugiere que una disminución de la serotonina puede resultar en depresión.
Sin embargo, las personas sin antecedentes personales o familiares de depresión generalmente no muestran cambios de humor significativos tras la depleción de triptófano, a pesar de que esta manipulación altera la actividad de regiones cerebrales reguladoras del estado de ánimo como la amígdala. Esto indica que la reducción de serotonina no induce depresión en todas las personas y que la vulnerabilidad individual juega un papel crucial. Un episodio depresivo previo podría alterar el sistema serotoninérgico, haciendo a la persona más vulnerable a futuros cambios en los niveles de serotonina. Además, incluso sin historia personal, una historia familiar de depresión puede aumentar la sensibilidad a la depleción de triptófano.
Genes y Vulnerabilidad: El Papel de la Genética
No se ha identificado un único gen o conjunto de genes que "cause" la depresión. En cambio, ciertas variaciones genéticas, llamadas polimorfismos, pueden aumentar el riesgo. Los genes influyen en la predisposición de muchas maneras, controlando el metabolismo de neurotransmisores, el número de neuronas y sinapsis, la transducción de señales intracelulares y la capacidad de adaptación a los estresores ambientales. El gen del transportador de serotonina es uno de los más estudiados. Contiene un polimorfismo con dos alelos: largo y corto. El alelo corto ralentiza la síntesis del transportador, lo que podría reducir la capacidad de adaptación de las neuronas serotoninérgicas al estrés. Las personas sanas con el alelo corto muestran una activación exagerada de la amígdala ante estímulos estresantes y pueden tener una mayor probabilidad de empeoramiento del humor tras la depleción de triptófano.
El estrés es un factor precipitante común para la depresión, pero no todos los expuestos al estrés se deprimen. Esto sugiere una interacción entre el estrés y la composición genética. Estudios han encontrado que los portadores del alelo corto del transportador de serotonina pueden ser especialmente vulnerables a la depresión bajo estrés, lo que se conoce como interacción gen-ambiente. Aunque no todos los estudios han observado esta interacción, se cree que el impacto de los genes individuales en el riesgo de depresión es pequeño. La interacción de múltiples genes y el estrés psicosocial en el riesgo de depresión es un área de investigación activa y compleja.
Factores de Crecimiento Neuronal: BDNF y la Plasticidad Cerebral
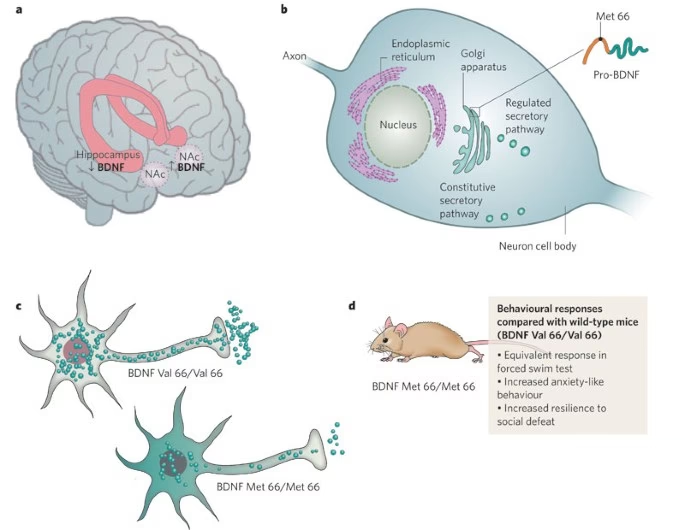
Un factor que puede moderar la interacción entre el polimorfismo del transportador de serotonina y el estrés psicosocial es el factor neurotrófico derivado del cerebro (BDNF, por sus siglas en inglés). Esta proteína es crucial para el nacimiento, supervivencia y maduración de las células cerebrales, y para la plasticidad sináptica a lo largo de la vida. El BDNF influye en estos procesos activando factores que estimulan la transcripción genética. Por ejemplo, en los núcleos del rafe, estimula genes relacionados con la función serotoninérgica, como el transportador de serotonina y la triptófano hidrolasa. A su vez, la activación de los receptores de serotonina estimula la expresión del gen del BDNF. Este ciclo promueve el crecimiento y la formación de sinapsis de las neuronas serotoninérgicas.
Un polimorfismo común en el gen del BDNF produce los alelos 'Val' y 'Met'. El alelo Met afecta el transporte y la secreción intracelular del BDNF. Las personas con el alelo Met tienden a tener un hipocampo más pequeño al nacer, actividad hipocampal alterada y memoria dependiente del hipocampo menos eficiente. Esto podría contribuir a una hipersensibilidad hipocampal al estrés. Algunos estudios sugieren que tener el alelo Met, junto con el alelo corto del transportador de serotonina y el estrés psicosocial (incluso en la infancia), aumenta la vulnerabilidad a la depresión más que los otros factores solos.
Además, se han encontrado bajos niveles de BDNF en el hipocampo y la corteza prefrontal de pacientes deprimidos sintomáticos en estudios postmortem. Los niveles séricos de BDNF también suelen ser anormalmente bajos en pacientes con trastorno depresivo mayor. En personas sanas, los niveles séricos de BDNF se correlacionan negativamente con la sensibilidad al estrés.
Adversidad Temprana y el Eje HPA: Cómo el Estrés Moldea el Cerebro
Los eventos estresantes, especialmente en la infancia, no ocurren al azar. El entorno de un niño está influenciado por su comportamiento y el de sus padres, con genes comunes que influyen en ambas generaciones. Esto dificulta separar los efectos genéticos de los ambientales en estudios humanos sobre la adversidad infantil y el riesgo de depresión adulta. Sin embargo, estudios experimentales con primates no humanos y roedores han proporcionado información valiosa. Por ejemplo, monos criados temporalmente por pares desarrollan respuestas de estrés exageradas asociadas con anomalías en la actividad serotoninérgica y en el eje hipotalámico-pituitario-adrenal (HPA).
Estudios en ratas sugieren que las experiencias infantiles pueden alterar la reactividad del eje HPA, mediado parcialmente por cambios epigenéticos en el gen del receptor de glucocorticoides. Estos cambios, que no implican alteraciones en la secuencia de ADN pero afectan la transcripción génica, aumentan la reactividad del eje HPA. Este proceso epigenético, influenciado por la serotonina, puede ocurrir incluso in utero y se cree que es de muy larga duración. Esto podría explicar por qué los pacientes con trastorno depresivo mayor a menudo muestran anormalidades en este sistema neuroendocrino.
El factor liberador de corticotropina (CRF) juega un papel clave en la sensibilidad al estrés. El estrés activa el hipotálamo (directamente) y la amígdala (indirectamente), liberando CRF y activando el eje HPA. El CRF también puede activar los sistemas de serotonina y norepinefrina. Las conexiones recíprocas entre el sistema de norepinefrina y el hipotálamo crean una cascada que activa progresivamente la señalización de CRF y norepinefrina, aumentando la vigilancia y el miedo. La desregulación combinada de los sistemas de CRF hipotalámico y extrahipotalámico podría explicar por qué los pacientes deprimidos a menudo tienen niveles inapropiadamente altos de CRF y norepinefrina, y muestran un procesamiento defectuoso de las amenazas ambientales y reacciones de estrés exageradas. La adversidad infantil puede contribuir a estas anomalías. Un estudio sugiere que el impacto del abuso infantil en la vulnerabilidad a la depresión puede ser moderado por polimorfismos en el gen del receptor de CRF tipo 1.
Otros Neurotransmisores Clave: Dopamina y Glutamato
Se cree que los cambios persistentes en la respuesta al estrés durante la infancia y la niñez aumentan la respuesta a eventos negativos de baja amenaza en la vida adulta, lo que podría explicar por qué los episodios depresivos recurrentes son más propensos a ocurrir independientemente del estrés. Además de la serotonina, la dopamina es cada vez más reconocida por su importante papel en la fisiopatología de la depresión mayor. Las amenazas ambientales percibidas por la amígdala aumentan los niveles de dopamina en la corteza prefrontal y el estriado ventral. Aunque normalmente hay un feedback inhibitorio para mantener la homeostasis, un estresor severo puede interrumpirlo alterando los niveles de BDNF en el estriado.
Este feedback anormal en el sistema dopaminérgico estriatal podría explicar por qué los pacientes deprimidos a menudo atribuyen una saliencia inapropiada incluso a estímulos ligeramente negativos. Además, los cambios en este sistema se cree que subyacen a la anhedonia, la incapacidad de sentir placer, reportada por muchos pacientes. La evidencia de anomalías dopaminérgicas en la depresión mayor es mixta, pero existe. Un polimorfismo en el gen del receptor de dopamina tipo 2 influye en el efecto de los eventos estresantes pasados sobre el estado de ánimo actual, sugiriendo otra interacción gen-ambiente.
El glutamato, un neurotransmisor aminoácido, también es vital para el crecimiento y la supervivencia celular. El estrés agudo aumenta moderadamente la neurotransmisión de glutamato sináptico, promoviendo el BDNF y la plasticidad. Sin embargo, el estrés crónico puede llevar a niveles excesivos de glutamato y la activación de receptores de glutamato tipo N-metil-D-aspártico (NMDA) fuera de la sinapsis. La hiperactivación de estos receptores aumenta el calcio intracelular a niveles tóxicos, disminuyendo el BDNF, induciendo atrofia y causando muerte celular. La degeneración de las células gliales, que normalmente ayudan a limpiar el exceso de glutamato, puede acelerar este proceso. Las interrupciones continuas en el sistema de glutamato pueden resultar en hipoactividad de regiones corticales. Estudios de neuroimagen en pacientes deprimidos han mostrado niveles reducidos de glutamato y proteínas asociadas en la corteza prefrontal, apoyando un papel para la toxicidad del glutamato en la fisiopatología.
Cambios Estructurales y Funcionales en el Cerebro Deprimido
Aunque tradicionalmente vista como una enfermedad con episodios seguidos de remisión, algunos pacientes muestran anormalidades neurobiológicas persistentes que pueden empeorar con el tiempo, llevando a la cronicidad. Similar a la esclerosis múltiple, podría haber patrones de progresión (recurrente-remitente vs. primario-progresivo). Estudios de neuroimagen estructural indican que individuos con episodios recurrentes pueden tener hipocampos relativamente pequeños incluso durante la remisión clínica. Se cree que la enfermedad recurrente o duradera y la falta de tratamiento antidepresivo contribuyen a reducciones progresivas del volumen del hipocampo, lo que podría explicar los problemas de memoria y otros síntomas. Los pacientes también pueden mostrar anomalías volumétricas en otras regiones subcorticales (amígdala, estriado ventral) y corticales (corteza cingulada anterior, corteza orbitofrontal, corteza prefrontal). Estas pueden persistir durante la remisión y contribuir a la sobrerreacción a estímulos amenazantes (reactividad cognitiva), aumentando el riesgo de recaída.
Las anormalidades estructurales se han relacionado con la función anormal del eje HPA. La hipercortisolemia crónica reduce los receptores de glucocorticoides en el hipocampo, lo que disminuye la transcripción de BDNF y genes diana, contribuyendo a la atrofia hipocampal. Esto, a su vez, puede comprometer aún más la función del eje HPA. Efectos similares mediados por el cortisol podrían explicar cambios en la amígdala y la corteza prefrontal. La toxicidad mediada por el glutamato también contribuye a la degeneración celular y la atrofia.
Las anomalías estructurales se vinculan a cambios en la actividad metabólica (anormalidades funcionales), que se asocian directamente con la gravedad de la enfermedad. En general, los estudios de neuroimagen indican múltiples anormalidades en la interconectividad de varias regiones subcorticales (límbicas) y corticales. Una relativa falta de regulación cortical sobre el sistema límbico ante la adversidad psicosocial podría explicar la sensibilidad al estrés, la labilidad emocional, la irritabilidad y la suicidalidad vistas en la depresión. Varias regiones cerebrales continúan mostrando anormalidades funcionales en la remisión clínica.
Estrategias de Tratamiento: Abordando la Base Neurológica
La complejidad de los sistemas involucrados explica por qué los tratamientos existentes, mayormente dirigidos a las monoaminas, a menudo no logran la remisión clínica. El retraso en la respuesta a los antidepresivos en quienes mejoran puede explicarse por sus efectos en el BDNF y otros sistemas reguladores del crecimiento. Estos efectos sobre la neuroplasticidad son relevantes, ya que el tratamiento podría revertir o prevenir anormalidades estructurales.
Nuevos fármacos investigados apuntan a los sistemas de CRF, dopamina y glutamato. Aunque un fármaco dirigido al receptor de CRF tipo 1 no mostró efecto significativo, el anestésico ketamina, que actúa sobre dopamina y glutamato, ha mostrado efectos antidepresivos robustos y rápidos incluso en depresión resistente, aunque con recaída frecuente en una semana. Otros agentes moduladores del glutamato como memantina y riluzol están en desarrollo. También hay interés en fármacos que actúan sobre GABA, melatonina y sustancia P.
La medicina personalizada basada en la farmacogenética busca adaptar el tratamiento según la composición genética del paciente. Un estudio encontró que el polimorfismo del gen del transportador de serotonina no afectaba la respuesta a citalopram, pero los portadores del alelo corto reportaron más efectos adversos. Sin embargo, un polimorfismo en el gen del receptor de serotonina tipo 2A sí influyó en el resultado del tratamiento con citalopram. Otros polimorfismos en genes del sistema serotoninérgico, el eje HPA, el sistema CRF y el sistema de norepinefrina, así como genes de enzimas hepáticas que metabolizan fármacos, también podrían afectar la respuesta al tratamiento.
Los enfoques no farmacológicos incluyen la psicoterapia, especialmente útil para quienes tienen antecedentes de adversidad infantil o estrés reciente. La terapia electroconvulsiva (TEC) es una opción para casos resistentes, aunque invasiva y con riesgo de amnesia. Nuevas técnicas de neuroestimulación, guiadas por la neurobiología, incluyen la estimulación del nervio vago (VNS), la estimulación magnética transcraneal (TMS) de la corteza prefrontal y la estimulación cerebral profunda (DBS) del cíngulo subgenual o el estriado ventral. La TMS se basa en la baja actividad prefrontal en pacientes deprimidos, y la DBS en disfunciones funcionales en el cíngulo subgenual/estriado ventral y anormalidades en la interconectividad corticolímbica. VNS y TMS están aprobadas en Canadá para depresión mayor, mientras que la DBS sigue siendo experimental. Estas técnicas, especialmente la TEC, se reservan para casos resistentes.
Preguntas Frecuentes
| Pregunta | Respuesta |
|---|---|
| ¿Es la depresión solo un problema de serotonina baja? | No, aunque la serotonina es importante, la base neurológica de la depresión es mucho más compleja, involucrando múltiples neurotransmisores, factores de crecimiento neuronal, el eje HPA y cambios estructurales/funcionales en diversas regiones cerebrales. |
| ¿La depresión es hereditaria? | No hay un único gen que cause la depresión, pero ciertas variaciones genéticas (polimorfismos) pueden aumentar la vulnerabilidad. El riesgo se relaciona más con la interacción de múltiples genes y factores ambientales como el estrés. |
| ¿El estrés en la infancia puede causar depresión en la adultez? | Sí, la adversidad psicosocial en la infancia es un factor de riesgo importante. Puede alterar sistemas cerebrales como el eje HPA a través de cambios epigenéticos duraderos, aumentando la vulnerabilidad al estrés y la depresión en la vida adulta. |
| ¿Por qué los antidepresivos tardan en hacer efecto o no funcionan para todos? | La mayoría de los antidepresivos actúan sobre las monoaminas, pero la fisiopatología de la depresión es más amplia. Su efecto no es inmediato porque implican cambios en la neuroplasticidad y otros sistemas reguladores del crecimiento, que toman tiempo. La variabilidad genética individual también influye en la respuesta al tratamiento. |
| ¿Hay cambios físicos visibles en el cerebro de una persona con depresión? | Sí, estudios de neuroimagen han mostrado reducciones de volumen (atrofia) en áreas como el hipocampo, la amígdala y la corteza prefrontal, especialmente en casos recurrentes o crónicos. También hay anormalidades en la actividad funcional y la conectividad entre regiones cerebrales. |
Conclusión: Hacia un Enfoque Multifacético
El trastorno depresivo mayor se entiende mejor como el resultado de la compleja interacción de múltiples factores genéticos heredados y la exposición a una amplia gama de variables ambientales a lo largo de la vida. Los roles exactos de los sistemas monoaminérgicos, otros neurotransmisores y sus objetivos a varios niveles continúan definiéndose. Las conexiones recíprocas entre regiones cerebrales y los mensajeros neuroquímicos involucrados se estudian cada vez más desde una perspectiva de sistemas.
Asimismo, se considera crecientemente el impacto del entorno psicosocial en la química, actividad y anatomía cerebral, y viceversa. El impacto de los eventos tempranos de la infancia, en particular, puede tener efectos duraderos, especialmente si implican cambios epigenéticos. Es crucial que tanto investigadores como clínicos presten atención a la posible contribución de la adversidad infantil a la psicopatología adulta, por ejemplo, incorporando la historia familiar y el contexto infantil en las entrevistas diagnósticas. Mucho queda por explorar sobre cómo los genes interactúan con otras variables ambientales, como las enfermedades inflamatorias o los factores estacionales, para influir en el riesgo de depresión. La depresión es una enfermedad con múltiples causas, tanto genéticas como ambientales, que requiere un enfoque multifacético en la investigación, el diagnóstico y el tratamiento.
Si quieres conocer otros artículos parecidos a La Base Neurológica de la Depresión puedes visitar la categoría Neurociencia.