En el vasto y complejo paisaje del cerebro, la depresión no es simplemente un estado de ánimo pasajero, sino un trastorno con profundas raíces neurológicas. Comprender qué la causa implica explorar desde los cambios más sutiles en la comunicación entre neuronas hasta las alteraciones en sistemas completos de neurotransmisores. A menudo, el término 'depresión' puede referirse tanto a un proceso celular específico que debilita las conexiones sinápticas (Depresión a Largo Plazo o LTD sináptica) como al trastorno clínico que afecta a millones de personas (Trastorno Depresivo Mayor o TDM). Es crucial distinguirlos, aunque la investigación sugiere que los mecanismos de plasticidad sináptica y los desequilibrios neuroquímicos, como los relacionados con el neurotransmisor GABA, están intrínsecamente ligados a la salud mental.
https://www.youtube.com/watch?v=0gcJCdgAo7VqN5tD
- La Depresión a Largo Plazo (LTD) Sináptica: Un Mecanismo de Plasticidad
- Trastorno Depresivo Mayor (TDM): El Papel Central de los Déficits GABAérgicos
- Modelos Animales: Ratones con Déficits GABAérgicos
- Comparación: TDM en Pacientes vs. Ratones γ2+/-
- Preguntas Frecuentes sobre la Depresión y el GABA
- Conclusiones y Perspectivas Futuras
La Depresión a Largo Plazo (LTD) Sináptica: Un Mecanismo de Plasticidad
La Depresión a Largo Plazo (LTD) es un hallazgo electrofisiológico experimental que representa lo opuesto a la Potenciación a Largo Plazo (LTP), el mecanismo celular asociado con el fortalecimiento sináptico y el aprendizaje. Mientras que la LTP se induce típicamente con estimulación de alta frecuencia o despolarización fuerte, la LTD ocurre cuando se aplica una energía más modesta, a menudo mediante estimulación de baja frecuencia. Este tipo de estimulación conduce a una activación menos intensa de los receptores NMDAR, lo que desencadena los procesos que llevan a la LTD.
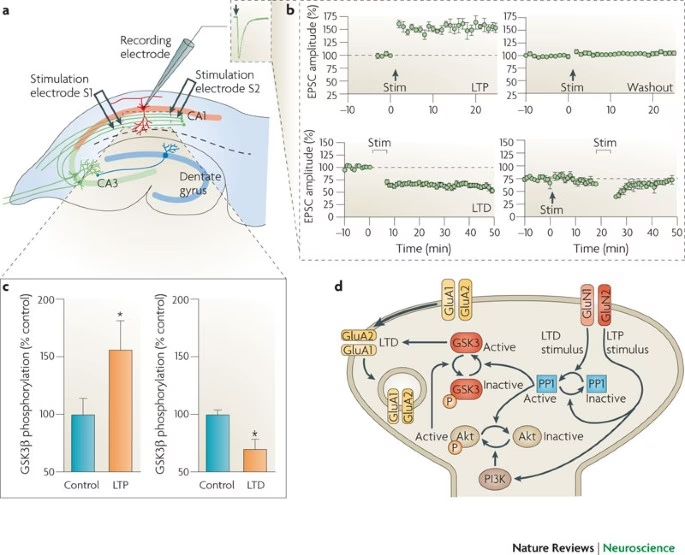
A nivel molecular, la inducción de LTD implica la reducción de la fuerza sináptica postsináptica. Una de las principales vías para lograr esto es a través de la endocitosis de receptores AMPAR. Mientras que la LTP promueve la inserción de subunidades AMPAR en la membrana postsináptica, la LTD induce su eliminación de la superficie celular. Esta eliminación reduce la capacidad de la sinapsis para responder a la liberación de neurotransmisores excitatorios, disminuyendo así la fuerza de la conexión.
Además de la endocitosis de AMPAR, la LTD puede estar relacionada con la reversión de las modificaciones sinápticas preexistentes. Si un tejido cerebral ya contiene numerosas "uniones inter-LINKed" (IPLs) formadas durante eventos de aprendizaje asociativo (representando fortalecimiento sináptico), la estimulación que induce LTD puede revertir algunas de estas IPLs. Los protocolos de estimulación de LTD, al causar una despolarización modesta, activan enzimas fosfatasas. Estas fosfatasas desfosforilan los receptores AMPAR, lo que contribuye a su endocitosis y a la subsiguiente reducción en el tamaño de las espinas dendríticas, revirtiendo así el fortalecimiento sináptico.
La LTD también puede implicar un mecanismo activo que reduce los potenciales postsinápticos. Esto puede ocurrir a través de la activación de sinapsis inhibitorias. En regiones corticales, por ejemplo, la inducción de LTP a menudo requiere reducir la inhibición GABAérgica. Esto sugiere que las sinapsis inhibitorias también se activan durante la estimulación. Si estas sinapsis inhibitorias se activan durante protocolos de LTD, la hiperpolarización resultante en las espinas dendríticas puede propagarse, causando una depresión neta en los potenciales que llegan al electrodo de registro, contribuyendo así a la LTD sináptica.
Trastorno Depresivo Mayor (TDM): El Papel Central de los Déficits GABAérgicos
Pasando del nivel sináptico a la complejidad de un trastorno clínico, el Trastorno Depresivo Mayor (TDM) es un síndrome neuropsiquiátrico complejo. Existe una creciente evidencia, tanto clínica como preclínica, que apunta a una asociación significativa y potencialmente causal entre el TDM y diversos tipos de déficits en la transmisión GABAérgica.
El GABA (ácido gamma-aminobutírico) es el principal neurotransmisor inhibitorio en el cerebro. Las neuronas GABAérgicas constituyen una proporción considerable de la población neuronal y son fundamentales para equilibrar y ajustar la neurotransmisión excitatoria en diversas regiones cerebrales. La hipótesis GABAérgica del TDM sugiere que las alteraciones en la transmisión GABAérgica representan aspectos fundamentalmente importantes en la secuencia etiológica de los trastornos depresivos mayores.
Evidencia Clínica de Déficits GABAérgicos en TDM
Los estudios en pacientes deprimidos han revelado consistentemente concentraciones reducidas del neurotransmisor GABA en diversas áreas cerebrales, detectadas mediante técnicas como la espectroscopía de resonancia magnética protónica. Se han observado reducciones significativas en la corteza occipital, la corteza cingulada anterior y la corteza prefrontal dorsomedial/dorsolateral de pacientes con TDM. Este fenotipo neuroquímico es coherente con la pérdida selectiva de interneuronas GABAérgicas en la corteza prefrontal dorsal observada en estudios postmortem. Los déficits de GABA parecen ser más pronunciados en subtipos de depresión más severos, como la depresión melancólica y resistente al tratamiento.
Más allá de las concentraciones del neurotransmisor, también se han encontrado alteraciones en la composición de las subunidades de los principales receptores que median la inhibición GABAérgica: los receptores GABA_A. Si bien la densidad total de sitios de unión a receptores GABA_A puede no estar alterada globalmente en cerebros postmortem de individuos deprimidos, la expresión de ARNm de diversas subunidades (como α1, α3, α4, δ, α5, β1, β3, γ2) muestra cambios descoordinados en diferentes regiones corticales y subcorticales implicadas en los trastornos del estado de ánimo. Estos cambios en la expresión de subunidades podrían dar lugar a subtipos de receptores GABA_A funcionalmente distintos, afectando la eficacia de la transmisión inhibitoria.
El Estrés como Factor de Vulnerabilidad y su Impacto en el Sistema GABAérgico
El estrés es reconocido como el factor de vulnerabilidad más importante para el TDM. La exposición al estrés, tanto en etapas tempranas del desarrollo como en la edad adulta, puede inducir un deterioro de la transmisión GABAérgica. Por ejemplo, el estrés por separación materna en roedores conduce a una reducción en la expresión de receptores GABA_A sensibles a benzodiazepinas en el cerebro adulto, asociada con una mayor reactividad al estrés. El estrés crónico en la edad adulta también puede reducir la abundancia y función de los receptores GABA_A, particularmente en la corteza cerebral y el hipocampo.
Una vía clave a través de la cual el estrés afecta la transmisión GABAérgica es a través de la modulación del eje hipotalámico-pituitario-adrenal (HPA). La hiperactivación de este eje, que resulta en una secreción aumentada de glucocorticoides, es un hallazgo común en subconjuntos de pacientes con TDM severo. Las regiones cerebrales que controlan el eje HPA, como el núcleo paraventricular del hipotálamo, están bajo un control inhibitorio GABAérgico. Los déficits de GABA en la corteza prefrontal y el hipocampo, inducidos por el estrés o por factores genéticos, pueden llevar a una hiperexcitabilidad local que se transmite al PVN, resultando en una hiperactivación del eje HPA. Además, el estrés crónico puede afectar la función GABAérgica directamente en el PVN.
Los neuroesteroides, cuya síntesis y niveles se ven alterados por el estrés, actúan como moduladores alostéricos de los receptores GABA_A, particularmente los subtipos extrasinápticos que contienen la subunidad δ. Neuroesteroides como la alopregnanolona (THP) y la alopregnanodiona (THDOC) pueden aumentar o reducir la inhibición tónica mediada por GABA en función del tipo celular y las condiciones. Las alteraciones en los niveles de estos neuroesteroides y en la regulación de los receptores α4βδ por el estrés y los ciclos hormonales se han implicado en trastornos como la depresión posparto (PPD) y el trastorno disfórico premenstrual (TDPM), destacando un posible factor de riesgo específico de sexo para el TDM.
Evidencia Genética y Farmacológica
La evidencia genética también apoya un papel para los déficits GABAérgicos. Polimorfismos genéticos en genes que codifican subunidades de receptores GABA_A se han asociado con trastornos afectivos como el trastorno bipolar y el TDM. Si bien estos polimorfismos pueden explicar solo una pequeña parte de la vulnerabilidad, sugieren que las alteraciones en la función de los receptores GABA_A pueden predisponer a estos trastornos.
Curiosamente, a pesar de que los tratamientos antidepresivos más utilizados (como los ISRS y los ATC) se diseñaron inicialmente para modular la transmisión monoaminérgica (serotonina, norepinefrina), la investigación sugiere que sus efectos terapéuticos podrían implicar, en última instancia, la potenciación de la transmisión GABAérgica. Estos fármacos pueden aumentar las concentraciones extracelulares de GABA en ciertas regiones cerebrales o modular indirectamente la actividad de las interneuronas GABAérgicas. Algunos antidepresivos también pueden actuar directamente como moduladores alostéricos positivos de los receptores GABA_A. Además, algunos fármacos que potencian la función GABAérgica, como el eszopiclone (un agonista alostérico selectivo de subtipos de receptores GABA_A), muestran promesa como antidepresivos.
Neurogénesis Hipocampal: Un Objetivo de los Antidepresivos
La neurogénesis adulta, particularmente en el giro dentado del hipocampo, es un proceso afectado en modelos animales de depresión y en pacientes crónicamente deprimidos (donde se observa reducción del volumen hipocampal). Este proceso de producción, maduración y supervivencia de nuevas neuronas es mejorado por los antidepresivos y parece ser necesario para muchos de sus efectos conductuales. La transmisión GABAérgica juega un papel esencial en el control de la neurogénesis adulta.
A diferencia de las neuronas maduras, donde el GABA suele tener un efecto hiperpolarizante, en los progenitores neuronales y neuronas inmaduras, la activación de los receptores GABA_A es despolarizante y excitatoria. Esta despolarización facilita la entrada de calcio y la activación de cascadas de señalización que promueven la maduración dendrítica y la supervivencia de estas neuronas inmaduras. El factor neurotrófico derivado del cerebro (BDNF), un objetivo importante de los antidepresivos, actúa aguas abajo de la señalización GABAérgica y también puede potenciar la liberación de GABA, creando un bucle de retroalimentación positiva crucial para la maduración neuronal.
Los déficits en la transmisión GABAérgica pueden afectar negativamente la supervivencia de las neuronas hipocampales recién nacidas. Modelos de ratón con déficits modestos en la subunidad γ2 del receptor GABA_A muestran no solo comportamientos ansiosos y depresivos, sino también una reducción en la supervivencia de las neuronas adultas generadas. Esto sugiere que la alteración de la neurogénesis, causada por déficits GABAérgicos, contribuye directamente a los fenotipos depresivos. La capacidad de los antidepresivos para potenciar la transmisión GABAérgica podría explicar, en parte, su efecto promotor sobre la neurogénesis y, por ende, su eficacia terapéutica.
Modelos Animales: Ratones con Déficits GABAérgicos
La evidencia más sólida de una relación causal proviene de modelos animales modificados genéticamente. Los ratones heterocigotos para la subunidad γ2 del receptor GABA_A (γ2+/-) presentan un déficit funcional moderado en los receptores GABA_A postsinápticos. Estos ratones exhiben una amplia gama de fenotipos que imitan síntomas clave de la depresión ansiosa melancólica en humanos, incluyendo aumento de la ansiedad, reducción del comportamiento de escape bajo estrés severo y efectos similares a la anhedonia (reducción del consumo de sacarosa).
Además de los fenotipos conductuales, los ratones γ2+/- muestran hiperactividad constitutiva del eje HPA (niveles elevados de corticosterona sérica) y una mayor sensibilidad conductual y endocrina a los antidepresivos, características también observadas en pacientes con depresión severa. Es notable que la inactivación selectiva del gen γ2 durante el desarrollo en el telencéfalo (incluyendo hipocampo y corteza frontal) es suficiente para inducir tanto la hiperactividad del eje HPA como los comportamientos ansioso-depresivos, sugiriendo que el déficit GABAérgico causal en estos ratones es extra-hipotalámico y se origina en etapas tempranas de la vida.
Los ratones knockout para la subunidad δ del receptor GABA_A, implicada en la inhibición tónica mediada por neuroesteroides, también muestran fenotipos relevantes. Específicamente, presentan déficits drásticos en la inhibición tónica GABAérgica en el período posparto, lo que se asocia con comportamiento ansioso, depresivo y alteraciones en el comportamiento materno, reflejando síntomas de la depresión posparto psicótica en humanos.
Estos modelos animales, al replicar aspectos celulares (déficits GABAérgicos, alteración de neurogénesis), neuroendocrinos (hiperactividad HPA), conductuales y farmacológicos (respuesta a antidepresivos) del TDM, proporcionan una fuerte evidencia de que los déficits en la transmisión GABAérgica pueden ser causales en la etiopatogenia de este trastorno.
Comparación: TDM en Pacientes vs. Ratones γ2+/-
La investigación en modelos animales, como los ratones γ2+/-, ha permitido establecer paralelismos notables con las características observadas en pacientes con Trastorno Depresivo Mayor. La siguiente tabla resume algunas de estas similitudes:
| Característica | Pacientes con TDM | Ratones GABA_A R γ2+/- |
|---|---|---|
| Déficits de GABA | Reducción en corteza, cingulado, occipital | Déficits estructurales/funcionales en corteza e hipocampo |
| Comorbilidad/Ansiedad | Alta comorbilidad con trastornos de ansiedad | Ansiedad elevada, inhibición conductual |
| Estrés temprano | Factor de riesgo etiológico importante | El fenotipo requiere déficits GABAérgicos en el desarrollo |
| Comportamiento de escape | Desesperanza, ideación suicida (en casos severos) | Comportamiento de escape reducido bajo estrés severo |
| Anhedonia | Síntoma central | Consumo de sacarosa reducido |
| Neurogénesis/Hipocampo | Reducción de volumen hipocampal (crónico) | Número reducido de neuronas maduras generadas |
| Eje HPA | Hiperactividad basal, aumento del cortisol | Aumento de la actividad basal del eje HPA |
| Respuesta a Antidepresivos | Mayor respuesta a ATC vs. ISRS en subtipos severos | Mayor sensibilidad conductual, desipramina eficaz, fluoxetina solo ansiolítica |
Preguntas Frecuentes sobre la Depresión y el GABA
Aquí respondemos algunas preguntas comunes basadas en la información presentada:
¿Qué es exactamente la Depresión a Largo Plazo (LTD) en neurociencia?
La LTD sináptica es un proceso de plasticidad neuronal que resulta en una disminución duradera en la fuerza de la conexión entre dos neuronas. Es lo opuesto a la potenciación a largo plazo (LTP) y se cree que juega un papel en procesos como el olvido o la eliminación de información irrelevante. Se induce con patrones de actividad neuronal específicos (baja frecuencia).
¿La LTD sináptica es lo mismo que el Trastorno Depresivo Mayor (depresión clínica)?
No, son conceptos distintos. La LTD sináptica es un mecanismo a nivel celular que altera la fuerza de una conexión neuronal. El Trastorno Depresivo Mayor es un trastorno clínico complejo que involucra alteraciones en múltiples circuitos cerebrales, sistemas de neurotransmisores (incluido el GABA), función neuroendocrina y neurogénesis. Si bien la plasticidad sináptica (incluida la LTD) probablemente contribuye a las disfunciones cerebrales observadas en el TDM, la LTD por sí sola no es la depresión clínica.
¿Cómo se relaciona el neurotransmisor GABA con la depresión clínica?
Existe una fuerte hipótesis de que los déficits en la transmisión GABAérgica contribuyen causalmente al TDM. Esto se basa en hallazgos de niveles reducidos de GABA en el cerebro de pacientes deprimidos y alteraciones en los receptores GABA_A. El GABA es crucial para equilibrar la actividad cerebral y su disfunción puede llevar a hiperexcitabilidad en circuitos clave implicados en el estado de ánimo y la respuesta al estrés.
¿El estrés puede causar déficits en el sistema GABAérgico?
Sí, el estrés, especialmente el crónico o el que ocurre en etapas tempranas de la vida, es un factor importante que puede alterar la expresión y función de los receptores GABA_A y afectar la transmisión GABAérgica, contribuyendo a la vulnerabilidad a la depresión.
¿Los medicamentos antidepresivos actúan sobre el sistema GABAérgico?
Aunque muchos antidepresivos se diseñaron inicialmente para aumentar los niveles de serotonina o norepinefrina, la evidencia sugiere que sus efectos terapéuticos finales pueden implicar la potenciación de la transmisión GABAérgica. Esto puede ocurrir indirectamente, a través de la modulación de interneuronas GABAérgicas, o incluso por efectos directos de algunos fármacos sobre los receptores GABA_A. Además, algunos fármacos que potencian el GABA, como ciertos moduladores de receptores GABA_A, muestran potencial antidepresivo.
¿Por qué es importante la neurogénesis hipocampal en el contexto de la depresión y el GABA?
La neurogénesis (la creación de nuevas neuronas) en el hipocampo está afectada en la depresión y es un objetivo de los antidepresivos. El GABA, sorprendentemente, tiene un efecto excitatorio en las neuronas inmaduras del hipocampo y es crucial para su maduración y supervivencia. Los déficits GABAérgicos pueden perjudicar este proceso neurotrófico, contribuyendo a la patología de la depresión. Los antidepresivos pueden promover la neurogénesis al potenciar la señalización GABAérgica en estas células inmaduras.
Conclusiones y Perspectivas Futuras
La investigación actual sugiere que los déficits en la transmisión GABAérgica, tanto a nivel sináptico como en circuitos más amplios, desempeñan un papel fundamental en la etiología del Trastorno Depresivo Mayor. El estrés crónico, los factores genéticos, las alteraciones en los neuroesteroides y la disfunción del eje HPA convergen para impactar negativamente el sistema GABAérgico, lo que a su vez afecta procesos clave como la neurogénesis hipocampal y la plasticidad sináptica.
Si bien la Depresión a Largo Plazo (LTD) sináptica es un mecanismo de plasticidad neuronal distinto del trastorno clínico, las alteraciones en la plasticidad sináptica en general son probablemente componentes de las disfunciones cerebrales subyacentes a la depresión. La evidencia de que los tratamientos antidepresivos, independientemente de su mecanismo de acción inicial, parecen converger en la potenciación de la transmisión GABAérgica refuerza la importancia de este sistema.
A pesar de los avances, quedan importantes interrogantes. Los mecanismos exactos que inician los déficits GABAérgicos aún no están completamente claros, y la forma en que estos déficits se relacionan con otras alteraciones observadas en la depresión (inflamación, disfunción glial) requiere más investigación. El desarrollo de terapias dirigidas específicamente a subtipos de receptores GABA_A o a circuitos GABAérgicos específicos implicados en el estado de ánimo podría ofrecer nuevas y más eficaces opciones de tratamiento para el TDM, abordando las causas subyacentes en lugar de solo los síntomas.
Si quieres conocer otros artículos parecidos a Depresión: GABA, Estrés y Plasticidad Sináptica puedes visitar la categoría Neurociencia.