En el vasto y complejo universo del cerebro humano, las proteínas desempeñan roles fundamentales para mantener la estructura, la comunicación y el funcionamiento adecuado de las neuronas. Una de estas proteínas, vital para la salud neuronal pero tristemente célebre por su implicación en diversas enfermedades neurodegenerativas, es la proteína tau.
Tau es un miembro destacado del grupo de Proteínas Asociadas a Microtúbulos (MAPs). Su naturaleza única reside en ser una proteína intrínsecamente desestructurada, lo que significa que carece de una estructura tridimensional fija y definida en su estado nativo. Esta característica le otorga una gran flexibilidad y una notable resistencia al calor y al tratamiento ácido, propiedades que la distinguen de muchas otras proteínas. Esta falta de estructura definida ha dificultado enormemente su análisis mediante técnicas tradicionales como la cristalografía, haciendo que la espectroscopia de resonancia magnética nuclear sea la herramienta principal para desentrañar sus conformaciones y movimientos.
- Estructura y Diversidad de Tau
- La Función Normal de Tau: Un Estabilizador Dinámico
- El Lado Oscuro de Tau: Misfolding y Agregación
- Modificaciones Postraduccionales de Tau
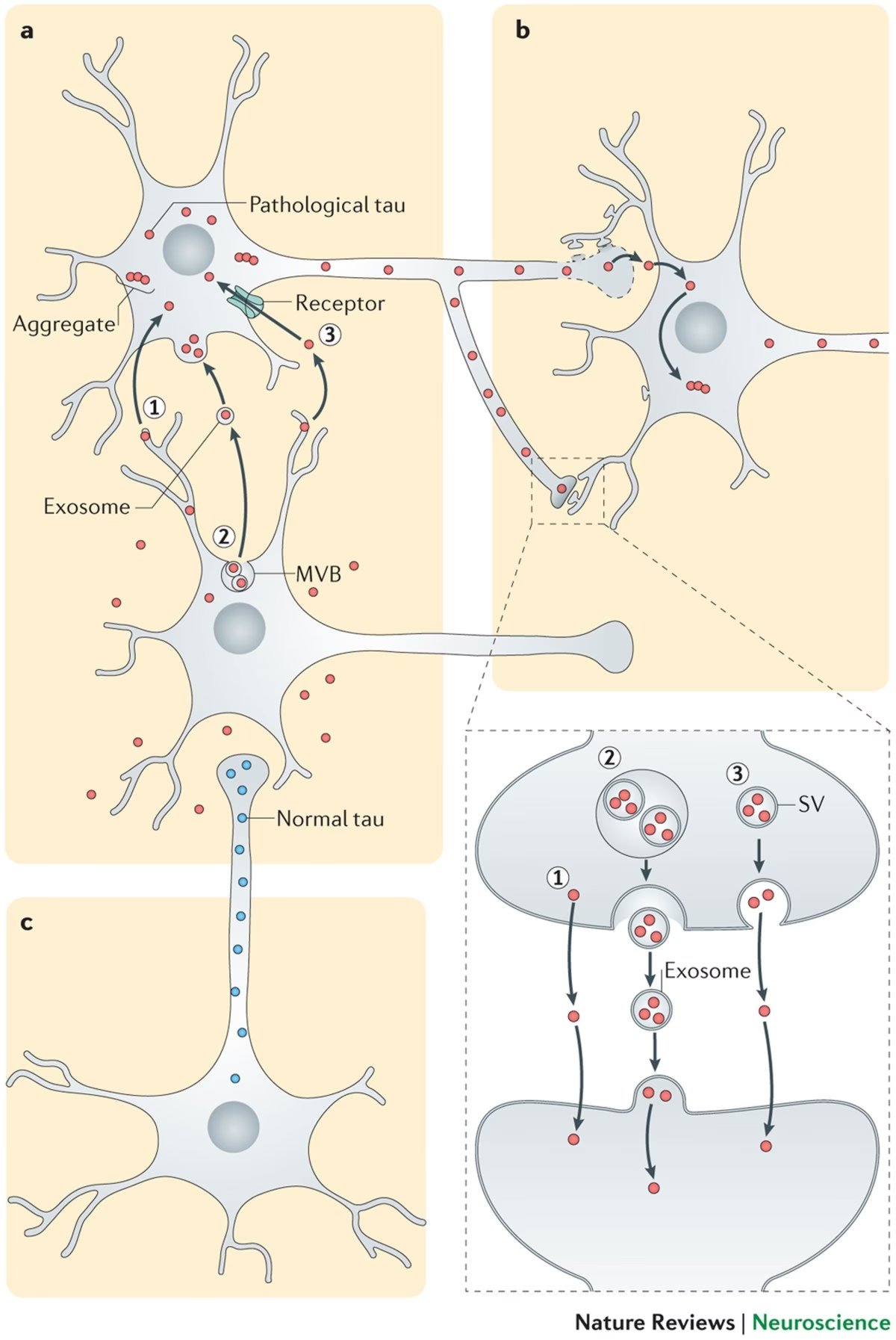
- Agregación y Propagación: Tau como Prion
- Tau y Enfermedades Neurodegenerativas (Tauopatías)
- Comparativa: Tau Sano vs. Tau Patológico
- Preguntas Frecuentes sobre la Proteína Tau
Estructura y Diversidad de Tau
El gen humano que codifica la proteína tau se localiza en el cromosoma 17, específicamente en la posición 17q21. Este gen consta de 16 exones, aunque no todos se traducen en la proteína final. Los exones constitutivos (1, 4, 5, 7, 9, 11, 12 y 13) están siempre presentes, mientras que los exones 2, 3 y 10 son objeto de empalme alternativo (alternative splicing) en el cerebro adulto.
Este proceso de empalme alternativo es crucial porque da lugar a la existencia de seis isoformas distintas de la proteína tau en el sistema nervioso central (SNC). La inclusión o exclusión del exón 10 es particularmente importante, ya que determina la presencia de un cuarto dominio de unión a microtúbulos. Así, se generan isoformas con tres repeticiones (3R tau, por exclusión del exón 10) o cuatro repeticiones (4R tau, por inclusión del exón 10) de estos dominios.
Las isoformas también varían en la presencia de inserciones en el extremo N-terminal, codificadas por los exones 2 y 3. El exón 2 puede estar presente solo, mientras que el exón 3 siempre aparece junto con el exón 2. Estas isoformas se conocen también como τ3L, τ3S, τ3, τ4L, τ4S y τ4, y su expresión varía significativamente a lo largo del desarrollo cerebral. Por ejemplo, durante las etapas fetales, predomina una única isoforma 3R sin inserciones N-terminales, mientras que en la edad adulta se expresan isoformas con una o dos inserciones N-terminales (derivadas de los exones 2 y/o 3) y con 3R o 4R.
Desde el punto de vista de su composición aminoacídica, tau presenta una estructura segmentada con cargas opuestas: un segmento N-terminal ácido, seguido de una región rica en prolina y una cola C-terminal básica. Los exones 2 y 3 contribuyen a la naturaleza ácida del extremo N-terminal, mientras que el exón 10 añade una secuencia cargada positivamente. La región N-terminal tiene un punto isoeléctrico (pI) de 3.8, el dominio rico en prolina de 11.4 y la región C-terminal de 10.8. Esta dualidad de cargas y la capacidad de sufrir múltiples modificaciones postraduccionales son clave para sus funciones fisiológicas.
Aunque tau se encuentra predominantemente en los axones de las neuronas, también puede detectarse en los oligodendrocitos, otro tipo de célula glial en el SNC. Otras proteínas asociadas a microtúbulos, como MAP2 y MAP4, muestran localizaciones diferentes: MAP2 se encuentra principalmente en el compartimento somatodendrítico de las neuronas, mientras que MAP4 tiene una distribución más amplia.
La Función Normal de Tau: Un Estabilizador Dinámico
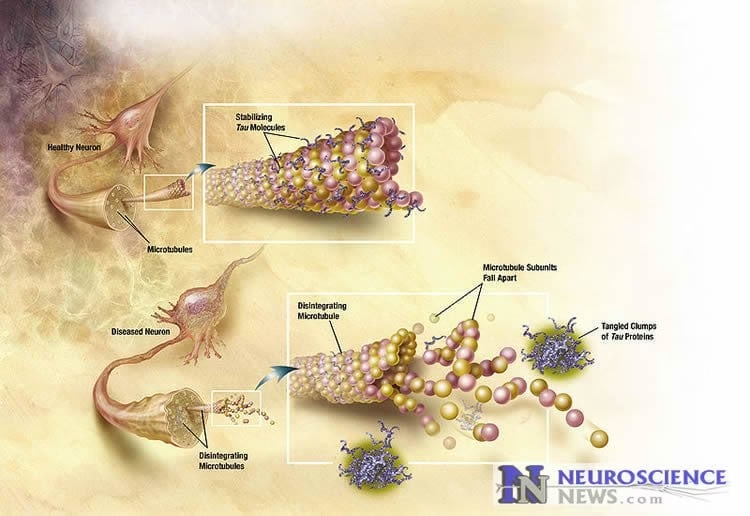
La función principal de la proteína tau en su estado normal es la de mantener la estabilidad de los microtúbulos. Los microtúbulos son filamentos proteicos grandes que forman parte del citoesqueleto celular, proporcionando estructura, organización y sirviendo como "autopistas" para el transporte de sustancias y orgánulos dentro de la célula. Tau se asocia a estos microtúbulos, promoviendo su ensamblaje y estabilidad, lo cual es esencial para procesos neuronales como el transporte axonal.
Recientemente, se ha descubierto un rol aún más dinámico para la proteína tau sana. Lejos de existir solo como moléculas individuales sueltas, tau puede autoasociarse de forma reversible, formando una especie de "envoltura" alrededor de los microtúbulos. Este comportamiento novedoso no solo sugiere que la autoasociación no siempre es patológica, sino que también parece tener un papel regulador. Al rodear los microtúbulos, tau puede influir en la capacidad de otras proteínas para unirse a ellos.
Además, esta envoltura de tau tiene la capacidad de compactar el microtúbulo. Los microtúbulos están formados por dímeros de tubulina, y existe una pequeña flexibilidad entre estos dímeros (aproximadamente 2 angstroms). La autoasociación de tau puede "exprimir" el microtúbulo, cambiando su compactación a lo largo de su longitud. Esto, a su vez, afecta cómo otras proteínas interactúan con él. Por ejemplo, se ha observado que la cinesina-1, una proteína motora que transporta cargas a lo largo de los microtúbulos, solo se une a microtúbulos expandidos, no compactados por tau.
Esta capacidad de compactación parece ser una característica evolucionada y específica, ya que otras MAPs, como MAP2 (también en neuronas), comparten esta habilidad, mientras que MAP4 (más ubicua) no la tiene. Esto sugiere que la regulación de la compactación de microtúbulos por tau y MAP2 podría ser particularmente importante para las funciones especializadas de las neuronas.
El Lado Oscuro de Tau: Misfolding y Agregación
Aunque en su estado normal tau tiene una baja propensión a plegarse incorrectamente o agregarse, ciertas alteraciones pueden desencadenar procesos patológicos. Cualquier cambio perjudicial, incluyendo una posible propagación tipo prión, puede inducir un plegamiento anormal y la formación de agregados, lo que lleva al desarrollo de enfermedades neurodegenerativas conocidas colectivamente como tauopatías.
Un evento clave en la patología de tau es la hiperfosforilación. La fosforilación es una modificación postraduccional normal y esencial para la función regulada de tau. Sin embargo, una fosforilación excesiva y aberrante conduce a la formación de tau hiperfosforilada (p-tau), que es la característica principal de diversas tauopatías. Este estado de hiperfosforilación provoca cambios conformacionales y de carga en la proteína, exponiendo los dominios de unión a microtúbulos y facilitando su oligomerización y autoagregación.
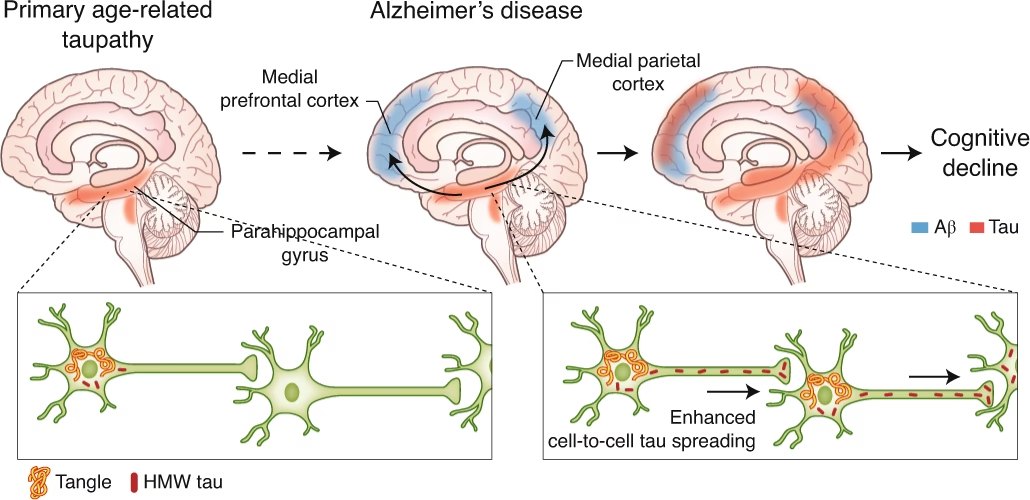
Con el tiempo, estos agregados de tau se ensamblan en estructuras filamentosas, primero en Filamentos Helicoidales Apareados (PHFs) y finalmente en las icónicas Ovillos Neurofibrilares (NFTs) que son un sello distintivo del cerebro en enfermedades como el Alzheimer (AD). La formación de estos agregados interrumpe el transporte de sustancias vitales a lo largo de los axones y desestabiliza los microtúbulos, comprometiendo la integridad estructural de la neurona.
En el cerebro de pacientes con AD, se observa una cantidad significativamente mayor (hasta cuatro veces más) de tau anormal o hiperfosforilada en comparación con individuos sanos. Esta tau malformada no solo pierde su función de estabilización de microtúbulos, sino que también adquiere una mayor tendencia a agregarse. La interacción defectuosa entre tau y los microtúbulos resultante afecta la plasticidad sináptica (la capacidad del cerebro para adaptarse y cambiar) y el transporte axonal, lo que contribuye al deterioro cognitivo asociado a la AD. Además, la fosforilación anormal de tau puede activar proteínas como la katanina, empeorando aún más los problemas en el ensamblaje de microtúbulos. La hiperfosforilación de tau es considerada un factor crítico en la progresión del daño cerebral relacionado con tau y está estrechamente ligada al desarrollo y avance de la AD, incluso más que la acumulación de la proteína beta-amiloide (Aβ) en algunas hipótesis.
Modificaciones Postraduccionales de Tau
La proteína tau es objeto de una regulación muy extensa a través de diversas modificaciones postraduccionales (PTMs) en sitios específicos de aminoácidos. Estas PTMs, que incluyen fosforilación, acetilación, metilación, sumoilación, ubiquitinación, truncamiento y glicación, impactan directamente sus características funcionales y su propensión a la agregación.
Fosforilación
Como se mencionó, la fosforilación es la PTM más común de tau, ocurriendo en residuos de serina, treonina y tirosina. La isoforma 2N4R de tau tiene hasta 85 posibles sitios de fosforilación. Está regulada por una compleja red de quinasas y fosfatasas. Las quinasas se clasifican en quinasas dirigidas a prolina (PDPKs) como GSK3β, CDK5 y MAPKs; quinasas no dirigidas a prolina (NonPDPKs) como TTBK1/2, CK1, PKA, PKB/Akt, etc.; y quinasas de tirosina como Src y Fyn.
Quinasas como GSK3β, CDK5, DYRK1A y CK1 están particularmente implicadas en la fosforilación anormal de tau en el cerebro con AD. Aproximadamente la mitad de los sitios de fosforilación de tau están seguidos por un residuo de prolina, lo que subraya la importancia de las PDPKs en su modificación patológica. Las fosfatasas como PP1, PP2A, PP2B, PP5 y PTEN son responsables de la desfosforilación, y su actividad o expresión a menudo se ve alterada en cerebros con AD.
La hiperfosforilación puede disociar tau de los microtúbulos e impedir su ensamblaje. El impacto exacto de la fosforilación en la agregación es complejo y a veces controvertido, con algunos sitios cuya fosforilación parece inhibir la agregación y otros que la aumentan.
Acetilación
La acetilación de residuos de lisina en tau está mediada por enzimas como p300/CREB binding protein (CBP). Los niveles de acetilación de tau están elevados en etapas tempranas y moderadas de tauopatía y contribuyen a la neurotoxicidad y el deterioro cognitivo. La acetilación reduce la afinidad de tau por los microtúbulos, perjudica el ensamblaje de tubulina y afecta su degradación. Su efecto en la agregación también es debatido, con estudios que sugieren tanto aceleración como inhibición.
Metilación
Se han identificado 11 sitios de mono- y dimetilación en tau. La tau metilada está presente en cerebros con AD y colocaliza con los NFTs en etapas tardías. La metilación tiene efectos mínimos en la afinidad por microtúbulos o el ensamblaje de tubulina, pero notablemente, atenúa drásticamente la propensión a la agregación, sugiriendo un papel protector.
Ubiquitinación y Sumoilación
Tau tiene una alta propensión a la ubiquitinación, lo que generalmente favorece su degradación por el proteasoma o autofagia. Sin embargo, los oligómeros de tau ubiquitinada pueden perjudicar la función del proteasoma. La ubiquitinación también puede inhibir el ensamblaje de microtúbulos y promover la agregación. La sumoilación, que afecta principalmente la interacción proteica y localización subcelular, se relaciona estrechamente con la fosforilación de tau. La sumoilación de tau promueve su hiperfosforilación, y viceversa, y puede inhibir la degradación de tau.
Truncamiento
En el cerebro con AD, tau es anormalmente truncada por diversas proteasas (caspasas, calpaína, AEP). La asparaginil endopeptidasa (AEP), cuya actividad aumenta con la edad, trunca tau en sitios como Asn255 y Asn368, generando fragmentos (ej. tau 1-368) más propensos a la fosforilación y agregación que la proteína completa. Estos fragmentos también pueden potenciar la producción de Aβ, propagando la patología de AD.
Modificación por DOPEGAL
El 3,4-Dihidroxifenilacetaldehído (DOPEGAL), un metabolito del norepinefrina, se acumula en neuronas con AD. DOPEGAL puede activar AEP y truncar tau, pero también puede modificar covalentemente residuos de lisina (ej. Lys353), desencadenando la agregación de tau y acelerando su patología y propagación.
Agregación y Propagación: Tau como Prion
La agregación patológica de tau sigue un mecanismo de nucleación-elongación. Los pasos iniciales implican la dimerización (nucleación), seguida por la adición rápida de monómeros a los extremos de los filamentos en crecimiento (elongación). Otros procesos como la fragmentación de filamentos y la nucleación secundaria también contribuyen. La fase de nucleación es lenta, mientras que la elongación es rápida. La adición de "semillas" preformadas (agregados pequeños) puede acelerar significativamente el proceso, reduciendo el tiempo de latencia.
Se ha documentado la "siembra cruzada" entre tau y otras proteínas mal plegadas, incluyendo Aβ, α-sinucleína e IAPP. La acumulación concurrente de Aβ y tau es un sello distintivo de la AD. Aβ puede acelerar la agregación de tau, aunque no se observa el mismo efecto de tau sobre Aβ. Estudios de PET han mostrado áreas de convergencia de patología Aβ y tau, sugiriendo una interacción física que impulsa la progresión de la enfermedad. La interacción Aβ-tau está ligada a la neurodegeneración y el deterioro cognitivo, indicando un papel sinérgico.
En subgrupos de pacientes con AD, así como en la Enfermedad de Parkinson y la Demencia con Cuerpos de Lewy (DLB), coexiste patología de tau y α-sinucleína. Tau y α-sinucleína interactúan directamente y se potencian mutuamente en su agregación y propagación. Los filamentos preformados (PFFs) de una proteína pueden inducir la agregación de la otra, tanto in vitro como in vivo.
Pacientes con diabetes mellitus tipo 2 (T2DM) tienen un mayor riesgo de AD. La T2DM se caracteriza por depósitos de polipéptido amiloide de islote (IAPP) en el páncreas. IAPP puede interactuar con tau, acelerando la formación de una "cepa" de tau más virulenta, con mayor actividad de siembra y neurotoxicidad.
La agregación de tau muestra similitudes con la de los priones, que adoptan conformaciones patológicas o "cepas" que se propagan y generan patrones neuropatológicos únicos. La evidencia sugiere que tau actúa como un prion. Las estructuras de los filamentos de tau extraídos de cerebros de pacientes con diferentes tauopatías, determinadas por crio-microscopía electrónica (crio-EM), revelan plegamientos únicos y específicos para cada enfermedad, que se conservan entre individuos con la misma patología.
En cerebros con AD, los inclusiones de tau tienen dos estructuras por crio-EM: PHFs y filamentos rectos, ambos con un núcleo ordenado común en forma de C (residuos 306–378 de la isoforma de 441 aa) y un "recubrimiento difuso" (fuzzy coat) en los extremos N y C-terminales. Estas estructuras pueden incorporar isoformas 3R y 4R. En otras tauopatías, las estructuras de los filamentos son distintas y explican su selectividad por ciertas isoformas. Por ejemplo, en la Enfermedad de Pick, los filamentos contienen predominantemente R1s y no R2s, lo que explica su selectividad por tau 3R. En la Degeneración Corticobasal (CBD) y la Parálisis Supranuclear Progresiva (PSP), los filamentos contienen R2s, explicando por qué solo contienen tau 4R.
Tau y Enfermedades Neurodegenerativas (Tauopatías)
Las enfermedades neurodegenerativas caracterizadas por la acumulación patológica de tau se denominan tauopatías. Estas incluyen, pero no se limitan a:
- Enfermedad de Alzheimer (AD): La tauopatía más común, caracterizada por ovillos neurofibrilares (NFTs) y placas de Aβ.
- Parálisis Supranuclear Progresiva (PSP): Acumulación de tau 4R.
- Degeneración Corticobasal (CBD): Acumulación de tau 4R.
- Enfermedad de Pick: Acumulación de tau 3R.
- Encefalopatía Traumática Crónica (CTE): Relacionada con traumatismos craneoencefálicos repetidos, presenta una tauopatía distintiva.
En todas estas condiciones, la disfunción y agregación de tau contribuyen a la neurodegeneración y al deterioro cognitivo y motor.
Comparativa: Tau Sano vs. Tau Patológico
| Característica | Tau Sano | Tau Patológico |
|---|---|---|
| Estructura Nativa | Intrínsecamente desestructurada, flexible | Hiperfosforilada, plegamiento anormal |
| Asociación a Microtúbulos | Estabiliza microtúbulos, promueve ensamblaje, puede compactarlos | Se disocia de microtúbulos, perjudica ensamblaje y transporte |
| Agregación | Baja propensión, autoasociación reversible | Alta propensión, forma oligómeros, PHFs, NFTs |
| Localización Primaria | Axones neuronales | Acumulación en somas, dendritas y axones; formación de inclusiones (NFTs) |
| Función Celular | Transporte axonal, plasticidad sináptica (indirecta), estructura citoesquelética | Neurotoxicidad, disfunción sináptica, muerte neuronal |
| PTMs Principales | Fosforilación regulada, otras PTMs normales | Hiperfosforilación aberrante, acetilación elevada, truncamiento patológico, etc. |
Preguntas Frecuentes sobre la Proteína Tau
¿Qué es la proteína tau?
La proteína tau es una proteína que se encuentra principalmente en las neuronas del cerebro, donde ayuda a estabilizar unas estructuras llamadas microtúbulos, que son esenciales para el transporte y la forma de las células neuronales.
¿Cuál es la función normal de tau en el cerebro?
En un cerebro sano, tau se une a los microtúbulos para darles soporte y estabilidad. Actúa como un "pegamento" que ayuda a mantener la integridad del citoesqueleto neuronal, permitiendo el transporte de nutrientes y otras moléculas a lo largo de los axones.
¿Cómo se relaciona tau con las enfermedades neurodegenerativas como el Alzheimer?
En enfermedades como el Alzheimer y otras tauopatías, la proteína tau se modifica anormalmente, principalmente por un proceso llamado hiperfosforilación. Esto hace que tau se separe de los microtúbulos y se agregue, formando ovillos neurofibrilares (NFTs) dentro de las neuronas. Estos agregados dañan las neuronas e interfieren con su función y supervivencia.
¿Qué son las tauopatías?
Las tauopatías son un grupo de enfermedades neurodegenerativas que se caracterizan específicamente por la acumulación patológica de la proteína tau en el cerebro. La Enfermedad de Alzheimer es la tauopatía más conocida, pero también incluyen la Parálisis Supranuclear Progresiva, la Degeneración Corticobasal, la Enfermedad de Pick y la Encefalopatía Traumática Crónica, entre otras.
¿La acumulación de tau es la única causa del Alzheimer?
Aunque la acumulación de tau patológica es un sello distintivo del Alzheimer y está estrechamente ligada al deterioro cognitivo y la muerte neuronal, el Alzheimer es una enfermedad compleja que también involucra la acumulación de otra proteína, beta-amiloide (Aβ), formando placas. La interacción entre tau y Aβ parece jugar un papel sinérgico en la progresión de la enfermedad, aunque algunas investigaciones sugieren que la patología de tau está más directamente relacionada con la neurodegeneración y los síntomas clínicos.
¿Puede la proteína tau propagarse de una neurona a otra?
Sí, la investigación sugiere que la proteína tau patológica puede comportarse de manera similar a un prion, propagándose de una neurona afectada a neuronas sanas vecinas. Este proceso de "siembra" y propagación contribuye a la extensión de la patología de tau por diferentes regiones del cerebro a medida que avanza la enfermedad.
En conclusión, la proteína tau es un componente esencial para la salud y función neuronal, manteniendo la estructura y el transporte a través de su asociación dinámica con los microtúbulos. Sin embargo, cuando sufre modificaciones patológicas, especialmente la hiperfosforilación, se convierte en un factor destructivo, agregándose y formando estructuras tóxicas que perturban la función celular y llevan a la neurodegeneración característica de las tauopatías. Comprender los mecanismos complejos que rigen la función normal de tau y las vías que conducen a su disfunción es fundamental para el desarrollo de terapias dirigidas a combatir estas devastadoras enfermedades.
Si quieres conocer otros artículos parecidos a Tau: Proteína Clave en Salud y Enfermedad Neuronal puedes visitar la categoría Neurociencia.