La Enfermedad de Parkinson (EP) es un trastorno neurodegenerativo complejo y progresivo que afecta predominantemente al sistema nervioso, impactando de manera significativa el movimiento y, con el tiempo, diversas funciones no motoras. Es el segundo trastorno neurodegenerativo más común después de la enfermedad de Alzheimer, y su prevalencia aumenta considerablemente con la edad.

Aunque la EP es ampliamente reconocida por sus síntomas motores característicos, como el temblor, la rigidez, la lentitud de movimiento (bradicinesia) y los problemas de equilibrio, su alcance va mucho más allá. Comprender la base neurológica de la EP es crucial para el desarrollo de terapias efectivas y para mejorar la calidad de vida de quienes la padecen.
- Las Bases Neuropatológicas de la Enfermedad de Parkinson
- La Patogénesis: Mecanismos Celulares Implicados
- Factores de Riesgo y Causas Potenciales
- El Sistema de Estadiaje de Braak: Un Marco para la Progresión
- Complicaciones Más Allá del Movimiento
- Modelos de Investigación para Entender la Enfermedad
- Preguntas Frecuentes sobre la Neurociencia del Parkinson
- Conclusión
Las Bases Neuropatológicas de la Enfermedad de Parkinson
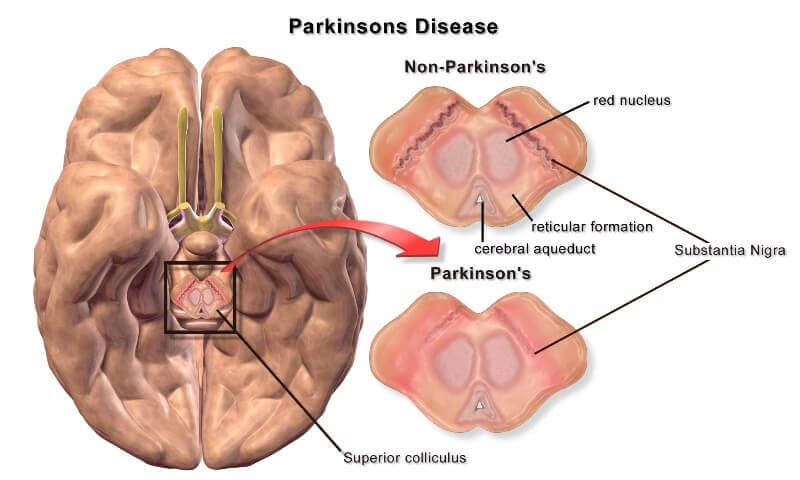
La característica patológica distintiva de la Enfermedad de Parkinson es la pérdida progresiva de neuronas específicas en una región del cerebro llamada sustancia negra pars compacta (SNpc). Estas neuronas son particularmente importantes porque producen un neurotransmisor vital: la dopamina. La disminución de dopamina en el cuerpo estriado, a donde proyectan estas neuronas, es lo que provoca los síntomas motores cardinales de la EP. Se estima que, al inicio de los síntomas motores, ya se ha producido una pérdida significativa (aproximadamente el 30%) de estas neuronas dopaminérgicas.
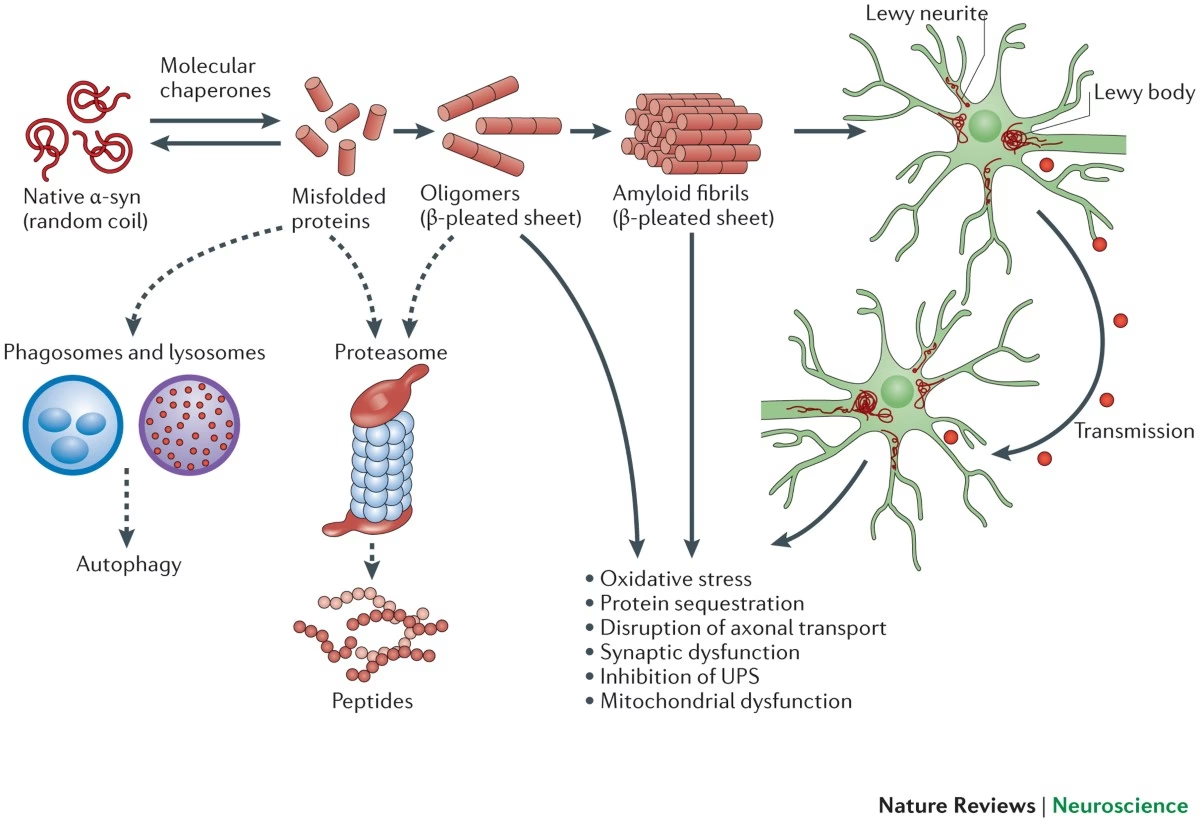
Además de la pérdida neuronal, otro sello distintivo de la EP es la presencia de acumulaciones anormales de proteínas dentro de las neuronas afectadas. Estas inclusiones se conocen como cuerpos de Lewy y neuritas de Lewy, y su componente principal es una proteína llamada alfa-sinucleína. La alfa-sinucleína es una proteína normalmente presente en las neuronas, especialmente en las terminales presinápticas, pero en la EP, se pliega de forma incorrecta y se acumula en agregados insolubles.
Aunque la pérdida de neuronas dopaminérgicas en la sustancia negra es central para los síntomas motores, la neurodegeneración en la EP no se limita a esta área. También se observa pérdida neuronal en otras regiones subcorticales, como el locus coeruleus (que produce norepinefrina), el núcleo basal de Meynert (colinérgico) y el bulbo olfatorio, entre otros. La afectación de estos otros sistemas de neurotransmisores se cree que contribuye a los síntomas no motores de la enfermedad.
Alfa-sinucleína y Cuerpos de Lewy
La proteína alfa-sinucleína juega un papel fundamental en la patología de la EP. En un cerebro sano, la alfa-sinucleína existe principalmente como una proteína desplegada o en formas tetraméricas solubles. Sin embargo, en la EP, adopta una estructura rica en hojas beta, propensa a la agregación. Estas formas mal plegadas se acumulan, inicialmente en oligómeros solubles (que se consideran altamente neurotóxicos) y posteriormente en fibrillas insolubles que forman el núcleo de los cuerpos de Lewy.
Los cuerpos de Lewy son inclusiones citoplasmáticas que varían en tamaño y morfología. Los cuerpos de Lewy clásicos del tronco encefálico tienen un núcleo denso rodeado por un halo, mientras que los cuerpos de Lewy corticales son más pequeños y carecen del halo distintivo. La distribución de los cuerpos de Lewy a lo largo del cerebro se utiliza para el sistema de estadiaje de Braak, que postula que la patología se extiende de forma secuencial desde las regiones inferiores del tronco encefálico y el bulbo olfatorio hasta la corteza cerebral en etapas avanzadas.
Es importante destacar que la patología de la alfa-sinucleína no se limita al cerebro. También se han encontrado depósitos de alfa-sinucleína fosforilada en el sistema nervioso periférico, incluyendo la médula espinal, los ganglios simpáticos e incluso en órganos periféricos como el tracto gastrointestinal, la piel y el corazón. Esta observación ha dado lugar a la "hipótesis del doble impacto" de Braak, que sugiere que la patología podría comenzar en sitios periféricos (como el intestino o el bulbo olfatorio) y luego propagarse al sistema nervioso central.
La Patogénesis: Mecanismos Celulares Implicados
La agregación de alfa-sinucleína es un evento central en la patogénesis de la EP, pero varios otros mecanismos celulares contribuyen a la disfunción y muerte neuronal.
Disfunción Mitocondrial
Las mitocondrias, las "centrales energéticas" de la célula, están implicadas de manera significativa en la EP. Se ha observado una deficiencia en la actividad del complejo I de la cadena de transporte de electrones mitocondrial en las neuronas de la sustancia negra de pacientes con EP. Esta deficiencia puede comprometer la producción de energía y aumentar el estrés oxidativo, dañando las células.
La exposición a ciertas toxinas ambientales, como el MPTP (1-metil-4-fenil-1,2,3,6-tetrahidropiridina) y pesticidas como la rotenona y el paraquat, que inhiben el complejo I, puede inducir un síndrome parkinsoniano en animales e incluso en humanos, proporcionando una fuerte evidencia del papel de la disfunción mitocondrial.
Además, varios genes asociados con formas familiares de EP, como PINK1 y Parkin (PARK6 y PARK2 respectivamente), están directamente involucrados en la mitigación, un proceso esencial para la eliminación selectiva de mitocondrias dañadas. Las mutaciones en estos genes pueden afectar la capacidad de la célula para deshacerse de las mitocondrias disfuncionales, lo que lleva a su acumulación y contribuye a la neurodegeneración. La alfa-sinucleína agregada también puede interactuar directamente con las mitocondrias, alterando su función.
Problemas en los Sistemas de Eliminación de Proteínas
Las células poseen sistemas sofisticados para eliminar proteínas dañadas o mal plegadas, principalmente el sistema ubiquitina-proteasoma (SUP) y la vía autofagia-lisosoma. El mal funcionamiento de estos sistemas se ha implicado en la EP, contribuyendo a la acumulación de alfa-sinucleína y otras proteínas tóxicas.
| Sistema de Eliminación | Mecanismo Principal | Implicación en EP |
|---|---|---|
| Sistema Ubiquitina-Proteasoma (SUP) | Marca proteínas anormales con ubiquitina para su degradación por el proteasoma. | Actividad reducida en el cerebro de pacientes con EP. Genes PARK (Parkin, UCH-L1) relacionados con su función. |
| Vía Autofagia-Lisosoma | Engulle componentes celulares (incluyendo proteínas) en vesículas (autofagosomas) que se fusionan con lisosomas para su degradación. Incluye macroautofagia, microautofagia y autofagia mediada por chaperonas (CMA). | Disfunción observada (acumulación de autofagosomas, disminución de proteínas lisosomales). Genes PARK (PINK1, Parkin, ATP13A2) y GBA1 (glucocerebrosidasa) implicados en esta vía o función lisosomal. |
La actividad reducida del SUP se ha reportado en el cerebro de pacientes con EP, y mutaciones en genes como Parkin (una ligasa E3 de ubiquitina) y UCH-L1 (una hidrolasa de ubiquitina) afectan directamente esta vía. De manera similar, se han encontrado anomalías en la vía autofagia-lisosoma, con evidencia de acumulación de autofagosomas y disminución de proteínas lisosomales clave. Mutaciones en genes como GBA1 (que codifica la enzima lisosomal beta-glucocerebrosidasa) son un factor de riesgo genético importante para la EP y resaltan el papel crítico de la función lisosomal.
Neuroinflamación
La inflamación en el sistema nervioso (neuroinflamación) es un componente cada vez más reconocido en la patogénesis de la EP. Se observa activación de células inmunes residentes del cerebro, llamadas microglía, así como infiltración de linfocitos T y un aumento en los niveles de citoquinas pro-inflamatorias en las áreas cerebrales afectadas de pacientes con EP.
Aunque inicialmente se pensó que la neuroinflamación era una respuesta secundaria al daño neuronal, ahora se cree que puede contribuir activamente a la progresión de la enfermedad. La alfa-sinucleína agregada puede activar directamente la microglía, perpetuando un ciclo de inflamación y daño neuronal. Factores genéticos relacionados con el sistema inmune (como la región HLA clase II) y estudios epidemiológicos que sugieren un menor riesgo en usuarios de antiinflamatorios no esteroideos (AINEs) como el ibuprofeno, apoyan aún más la participación de la neuroinflamación.
Factores de Riesgo y Causas Potenciales
La causa exacta de la EP es desconocida, pero se considera una enfermedad multifactorial que resulta de una compleja interacción entre factores genéticos y ambientales.
- Edad: Es el factor de riesgo más importante. La incidencia de la EP aumenta drásticamente con la edad, siendo más común su inicio después de los 60 años. Las formas de inicio temprano (antes de los 50) son menos frecuentes y a menudo están más relacionadas con factores genéticos específicos.
- Genética: Aunque la mayoría de los casos de EP son esporádicos (sin historia familiar clara), aproximadamente el 10-15% tienen antecedentes familiares, y un pequeño porcentaje (5-10%) son causados por mutaciones en genes específicos (formas monogénicas o familiares). Se han identificado más de 20 genes (llamados genes PARK) asociados con la EP. Mutaciones en SNCA (alfa-sinucleína) y LRRK2 son causas comunes de formas autosómicas dominantes, mientras que mutaciones en PRKN (Parkin), PINK1 y DJ-1 se asocian con formas autosómicas recesivas, a menudo de inicio más temprano. Además, se han identificado numerosos factores de riesgo genéticos, siendo las mutaciones en el gen GBA1 (glucocerebrosidasa) uno de los más importantes numéricamente, aumentando el riesgo de desarrollar EP.
- Factores Ambientales: La exposición a ciertas toxinas ambientales ha sido implicada, aunque la evidencia es compleja. La exposición al MPTP, un subproducto de drogas ilegales, causa parkinsonismo. Pesticidas como el paraquat y la rotenona, que son inhibidores del complejo I mitocondrial, también se han asociado con un mayor riesgo en algunos estudios. Otros factores como la exposición a metales pesados o el agua de pozo han sido investigados, pero su papel no está claro. Curiosamente, se ha observado una relación inversa entre el tabaquismo y el consumo de cafeína con el riesgo de EP en muchos estudios, lo que sugiere un posible efecto protector, aunque los mecanismos exactos y la causalidad directa siguen siendo objeto de debate.
El Sistema de Estadiaje de Braak: Un Marco para la Progresión
Basado en el examen postmortem de cerebros, el neuropatólogo Heiko Braak propuso un sistema de estadiaje para describir la progresión de la patología de los cuerpos de Lewy en la EP. Este sistema describe seis etapas, comenzando en las regiones inferiores del tronco encefálico (núcleos motores del vago, bulbo olfatorio) en las etapas 1 y 2, donde predominan los síntomas no motores (estreñimiento, anosmia, trastornos del sueño REM). La patología luego asciende a la sustancia negra y el locus coeruleus (etapa 3), momento en el que suelen aparecer los síntomas motores. En las etapas posteriores (4-6), la patología se extiende a las regiones límbicas (amígdala, corteza transentorrinal) y finalmente a la neocorteza, asociándose con síntomas más severos, problemas cognitivos y demencia.
Aunque influyente, el sistema de Braak ha generado controversia. No todos los cerebros de pacientes con EP siguen este patrón, y la correlación entre el estadiaje de Braak y la severidad de los síntomas clínicos no siempre es consistente. Sin embargo, subraya la naturaleza progresiva y multifocal de la enfermedad.
Complicaciones Más Allá del Movimiento
La EP es mucho más que un trastorno motor. A medida que avanza la enfermedad, muchos pacientes desarrollan una amplia gama de síntomas no motores que pueden tener un impacto significativo en su calidad de vida y a menudo son menos sensibles a la terapia dopaminérgica.
Estos síntomas incluyen:
- Problemas cognitivos y demencia (especialmente en etapas avanzadas, a menudo con afectación de funciones ejecutivas, atención y memoria de trabajo).
- Alteraciones del estado de ánimo, como depresión y ansiedad.
- Trastornos del sueño, incluyendo el trastorno de conducta del sueño REM (actuar los sueños).
- Síntomas autonómicos, como estreñimiento, problemas urinarios y cambios en la presión arterial (hipotensión ortostática).
- Pérdida del sentido del olfato (anosmia), que a menudo aparece en las primeras etapas o incluso antes de los síntomas motores.
- Fatiga, dolor y disfunción sexual.
La aparición de demencia en la EP puede estar relacionada no solo con la patología de la alfa-sinucleína en la corteza, sino también con la coexistencia de patología de otras proteínas, como la proteína tau (característica del Alzheimer) y las placas de beta-amiloide. La interacción entre estas diferentes proteinopatías puede contribuir a la heterogeneidad clínica de la enfermedad.
Modelos de Investigación para Entender la Enfermedad
El estudio de la EP se beneficia enormemente de modelos de investigación, tanto in vitro (cultivos celulares, células madre pluripotentes inducidas por humanos - iPSCs) como in vivo (modelos animales).
Los modelos basados en iPSCs derivadas de pacientes con EP permiten estudiar los mecanismos de la enfermedad en tipos neuronales humanos relevantes (como las neuronas dopaminérgicas) en una placa de cultivo ("Parkinson en una placa"). Esto facilita el estudio de la agregación de proteínas y la búsqueda de fármacos.
Los modelos animales, particularmente roedores y primates no humanos, son cruciales para estudiar la progresión de la enfermedad y probar terapias potenciales. Existen modelos genéticos (basados en mutaciones en genes PARK) y modelos basados en toxinas (como 6-OHDA o MPTP). Si bien los modelos genéticos pueden replicar aspectos moleculares y de agregación proteica, a menudo no reproducen completamente la pérdida neuronal y los síntomas motores típicos de la EP esporádica.
Los modelos basados en toxinas, especialmente el modelo de MPTP en primates no humanos, replican de manera más cercana la pérdida de neuronas dopaminérgicas y los síntomas motores observados en humanos, aunque no siempre desarrollan cuerpos de Lewy clásicos. El desarrollo de modelos que repliquen mejor los síntomas no motores, como los déficits cognitivos, sigue siendo un área activa de investigación.
Preguntas Frecuentes sobre la Neurociencia del Parkinson
¿Qué causa la Enfermedad de Parkinson?
La causa exacta de la EP es desconocida. Se cree que es una enfermedad multifactorial que resulta de una compleja interacción entre la predisposición genética y la exposición a ciertos factores ambientales a lo largo de la vida.
¿Es el Parkinson una enfermedad genética?
La mayoría de los casos de Parkinson son esporádicos (sin una causa genética única obvia). Sin embargo, un porcentaje menor (5-10%) son causados por mutaciones en genes específicos (formas familiares). Además, se han identificado numerosos genes que aumentan el riesgo de desarrollar la enfermedad, actuando como factores de riesgo.
¿Qué son los cuerpos de Lewy?
Los cuerpos de Lewy son acumulaciones anormales de proteínas que se forman dentro de las neuronas en el cerebro de las personas con Enfermedad de Parkinson y otras enfermedades relacionadas. Están compuestos principalmente por la proteína alfa-sinucleína que se ha plegado y agregado de forma incorrecta.
¿Por qué la pérdida de neuronas en la sustancia negra es tan importante?
Las neuronas de la sustancia negra producen dopamina, un neurotransmisor esencial para controlar el movimiento. La pérdida de estas neuronas reduce drásticamente la cantidad de dopamina en el cerebro, lo que lleva a los síntomas motores característicos de la EP, como la lentitud, la rigidez y el temblor.
¿Existe una cura para la Enfermedad de Parkinson?
Actualmente, no existe una cura para la Enfermedad de Parkinson. Los tratamientos disponibles se centran principalmente en aliviar los síntomas (como la terapia de reemplazo de dopamina) y mejorar la calidad de vida. La investigación en curso busca desarrollar terapias que puedan frenar o detener la progresión de la enfermedad.
Conclusión
La Enfermedad de Parkinson es un desafío complejo para la neurociencia. La pérdida de neuronas dopaminérgicas y la agregación de alfa-sinucleína en los cuerpos de Lewy son características centrales, pero la patogénesis implica una red intrincada de disfunción celular, incluyendo problemas mitocondriales, fallos en la eliminación de proteínas y neuroinflamación. Factores genéticos y ambientales contribuyen a la susceptibilidad individual. La investigación continua, utilizando modelos avanzados y enfoques multidisciplinarios, es fundamental para desentrañar completamente los mecanismos subyacentes y desarrollar tratamientos que no solo controlen los síntomas, sino que también modifiquen el curso de esta devastadora enfermedad.
Si quieres conocer otros artículos parecidos a Parkinson: La Neurociencia Detrás puedes visitar la categoría Neurociencia.