La terapia génica, con su doble promesa de abordar la etiología de la enfermedad y ofrecer una corrección a largo plazo, está experimentando un resurgimiento significativo. Esta aproximación terapéutica es particularmente atractiva para las enfermedades neurodegenerativas, donde las estrategias farmacológicas convencionales a menudo han resultado decepcionantes. El éxito reciente de una terapia génica basada en vectores virales para la Atrofia Muscular Espinal (SMA) —promoviendo la supervivencia y la función motora con una sola inyección— ofrece un paradigma para este tipo de intervención terapéutica y una plataforma sobre la cual construir.
Si bien persisten desafíos, el optimismo renovado se deriva en gran medida de los avances en el desarrollo de vectores virales que pueden administrar genes de manera difusa en todo el sistema nervioso central (SNC), así como de herramientas de ingeniería genómica que pueden manipular las vías de la enfermedad de formas antes imposibles. La Atrofia Muscular Espinal (SMA) es sin duda un caso de estudio ejemplar en este campo.
Terapia Génica para la Atrofia Muscular Espinal: Un Caso de Éxito
La Atrofia Muscular Espinal (SMA) es un trastorno neuromuscular autosómico recesivo causado por mutaciones en el gen que codifica la proteína de la neurona motora de supervivencia (SMN), lo que lleva a una pérdida de células del asta anterior en la médula espinal y resulta en debilidad progresiva de las neuronas motoras inferiores. Más del 95% de los casos se deben a deleciones homocigotas en el exón 7 del gen SMN1. Sin embargo, la gravedad del fenotipo está influenciada por el número de copias del gen SMN2, una copia casi idéntica del SMN1 que codifica una proteína idéntica. No obstante, SMN2 produce principalmente una proteína truncada porque solo el 10% de sus transcritos se traducen completamente debido al empalme alternativo del exón 7 y la subsiguiente degradación del pre-ARNm. La gravedad de la enfermedad varía desde la SMA tipo I de inicio temprano y severo hasta la SMA tipo IV de inicio en la edad adulta y progresión lenta, y está inversamente relacionada con el número de copias del gen SMN2.
Actualmente, dos terapias génicas están aprobadas para la SMA: nusinersen (un ASO administrado por vía intratecal) y onasemnogene abeparvovec (una terapia génica basada en un vector AAV administrado por vía intravenosa). Nusinersen es un oligonucleótido antisentido (ASO) complementario a un elemento regulador aguas arriba del exón 7 en SMN2, lo que lleva a su inclusión durante el empalme alternativo y produce un producto de ARNm estable. Onasemnogene abeparvovec es una terapia génica basada en un vector AAV9, que contiene una copia funcional completa del gen SMN1, aprobada para el tratamiento de niños con SMA menores de 2 años.
Además de estas terapias génicas, la SMA también cuenta con un fármaco modificador del empalme de molécula pequeña aprobado, risdiplam, que está autorizado para el tratamiento en todas las edades y subtipos de SMA. No se han realizado comparaciones directas entre estas tres terapias. Dado que existe una discapacidad residual con cualquiera de los agentes individuales y el potencial de un efecto sinérgico, hay mucho interés en enfoques combinatorios, con la publicación de datos observacionales y ensayos clínicos en curso.
Consideraciones de Tratamiento y Seguimiento Post-Tratamiento
Nusinersen se administra por vía intratecal con 4 dosis de carga dentro de los primeros 2 meses, seguidas de dosis de mantenimiento cada 4 meses. Se recomiendan hemogramas completos y estudios de coagulación al inicio y antes de administrar cada dosis, y se debe monitorear clínicamente a los pacientes para detectar sangrado. De manera similar, aunque rara, se ha observado toxicidad renal, incluido el riesgo de glomerulonefritis grave, con tratamientos con ASO. Las recomendaciones actuales sugieren obtener una prueba cuantitativa de proteína en orina puntual al inicio y antes de administrar cada dosis como estrategia de mitigación de riesgos. Los riesgos de seguridad relacionados con la vía de administración intratecal incluyen dolores de cabeza post-punción lumbar, dolor de espalda, infección y sangrado. Se han reportado meningitis infecciosa y aséptica, aracnoiditis e hidrocefalia después de la comercialización. Los riesgos de seguridad clave relacionados con el ASO en sí incluyen trombocitopenia, anomalías de la coagulación y toxicidad renal. La trombocitopenia es una complicación bien descrita de los ASO y puede variar desde una forma transitoria, leve y asintomática hasta una forma grave rara asociada con hemorragias intracraneales.
Onasemnogene abeparvovec se administra como una infusión intravenosa única. Antes del tratamiento, se deben evaluar los títulos de anticuerpos anti-AAV9, ya que no hay datos que informen sobre la seguridad o eficacia en pacientes con títulos elevados (>1:50). En los ensayos clínicos, se observaron elevaciones asintomáticas en el 90% de los pacientes, generalmente comenzando alrededor del día 7 y resolviéndose a los 2 meses después del tratamiento mientras se seguía un régimen de esteroides. Sin embargo, en los datos posteriores a la comercialización, se han reportado casos sintomáticos graves de lesión hepática, incluidos algunos casos de insuficiencia hepática y muerte. Por lo tanto, los pacientes con niveles de transaminasas o bilirrubina total superiores al doble del límite superior normal no deben recibir la dosis, excepto en casos de hiperbilirrubinemia neonatal aislada. Según las recomendaciones actuales, los pacientes deben recibir un ciclo de esteroides comenzando 24 horas antes de la infusión y continuarlo durante al menos 1 mes después de la dosificación. Las pruebas de función hepática y los parámetros de coagulación deben ser monitoreados de cerca durante este tiempo. Dado el uso de esteroides, se debe considerar ajustar el calendario de vacunación. Los riesgos de seguridad clave de onasemnogene abeparvovec como terapia génica con vector AAV incluyen hepatotoxicidad, trombocitopenia, microangiopatía trombótica (MAT) y eventos cardíacos. Se cree que la hepatotoxicidad con elevación de transaminasas es secundaria a la inmunidad mediada por células contra la cápside del vector.
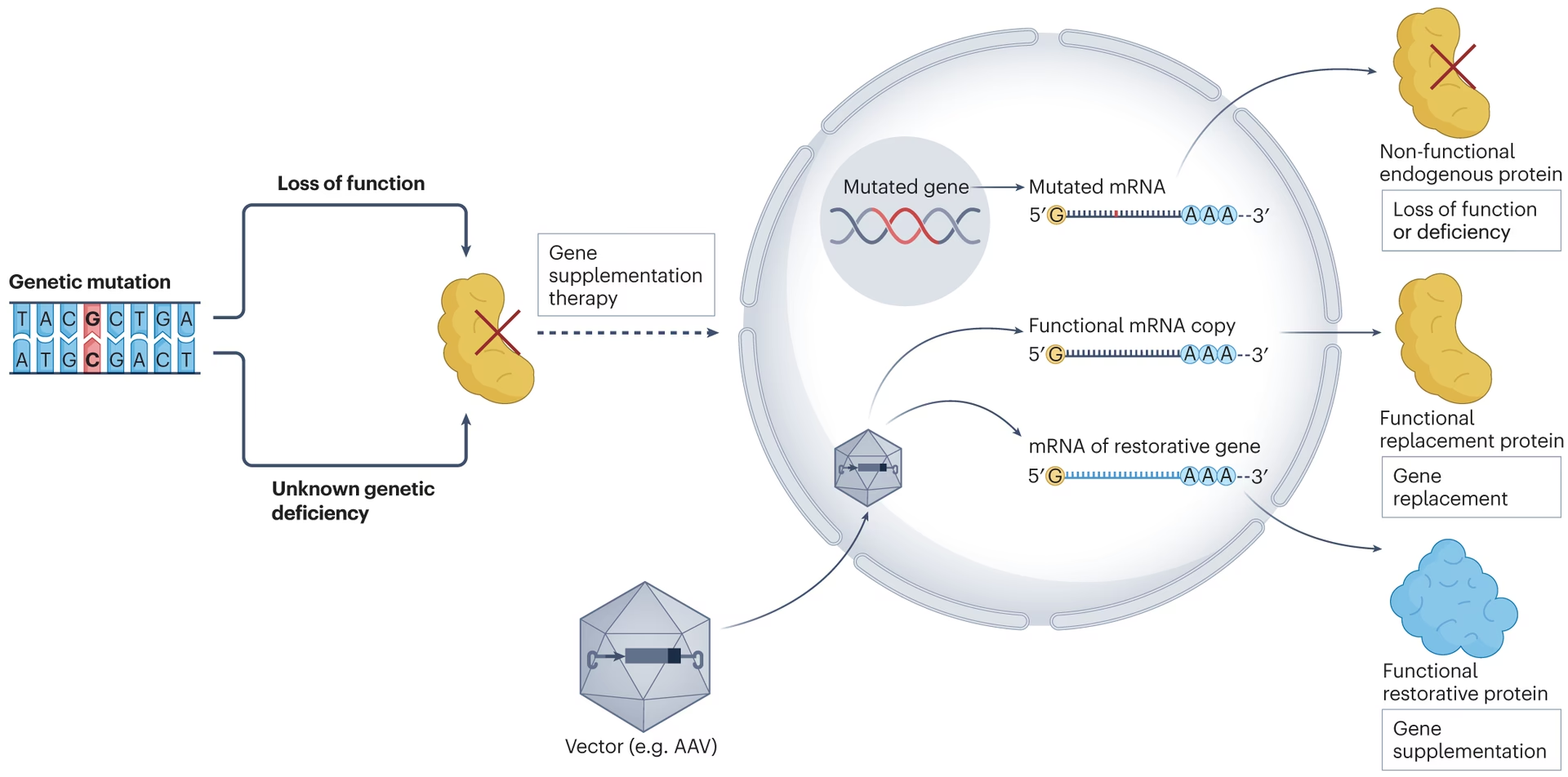
Tipos de Terapia Génica
La idea de que un gen puede ser entregado a tipos celulares específicos y que su expresión puede llevar a una eficacia terapéutica, mejorando drásticamente la calidad de vida de los pacientes, fue introducida hace más de 40 años. En este contexto, el fármaco, que en el caso de la terapia génica es un gen, se empaqueta dentro de un vector utilizado para facilitar su entrada en las células del paciente. La noción de terapia génica ha evolucionado, y en general, nos referimos a terapia génica cuando un proceso terapéutico implica la manipulación genética de las células del paciente mediante el uso de un ácido nucleico.
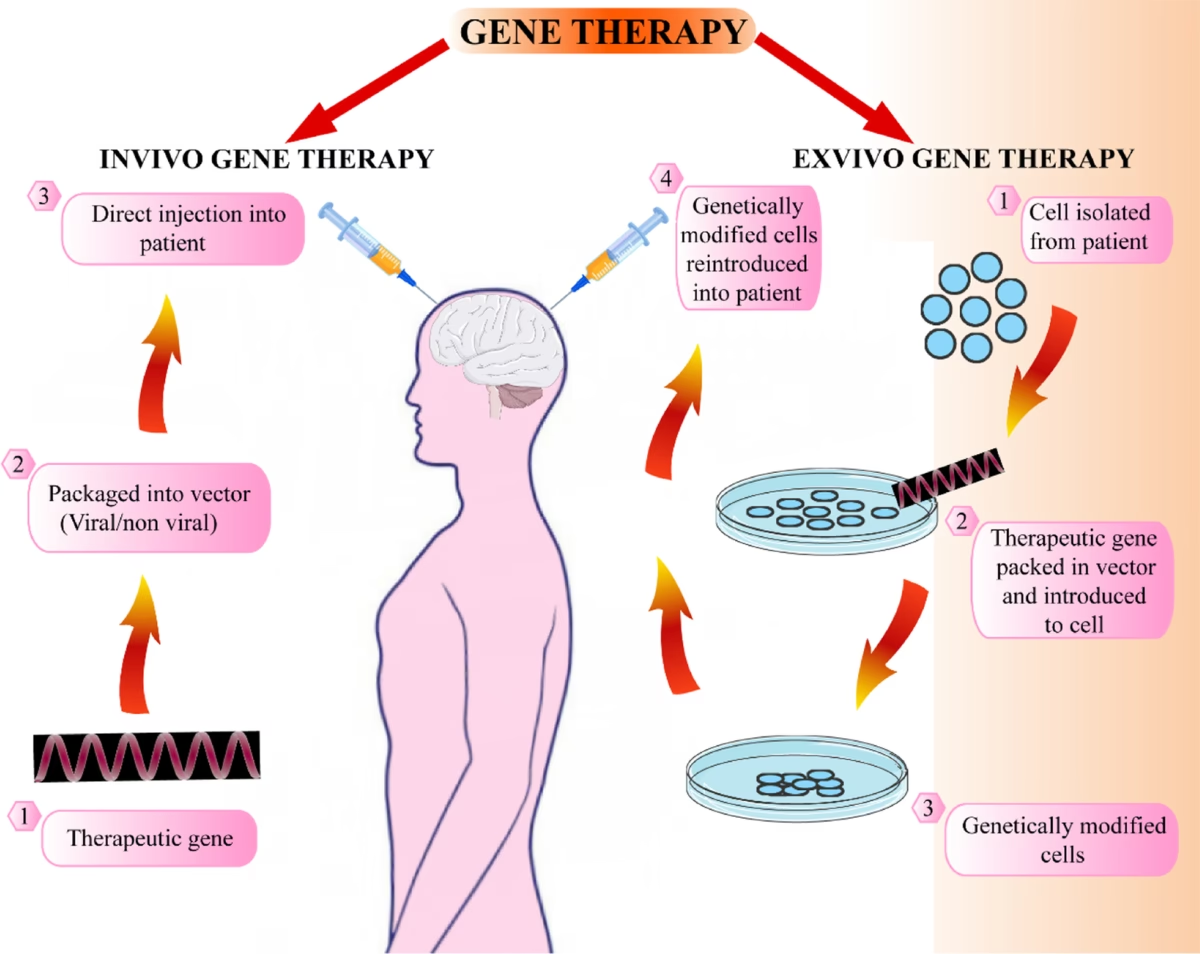
Existen básicamente tres tipos de terapia génica: ex vivo, in vivo e in situ. En la terapia génica ex vivo, las células objetivo se extraen del cuerpo del paciente, se modifican genéticamente mediante la adición del gen terapéutico u otras manipulaciones genéticas que permiten la corrección del fenotipo de la enfermedad. Las células 'corregidas' se reinfunden posteriormente al paciente. Este tipo de intervención también se denomina terapia génica in vitro y es particularmente aplicable a las enfermedades de la sangre, utilizando células madre hematopoyéticas (CMH) o células T (como en las terapias CAR-T). Los vectores virales utilizados suelen ser retrovirales o lentivirales.
Si el órgano objetivo es el cerebro, el canal espinal o el hígado, se emplea otro tipo de terapia, denominada terapia génica in vivo. En este entorno, el vector terapéutico se administra sistémicamente en la circulación sanguínea o el líquido cefalorraquídeo del paciente, y dependiendo de la enfermedad, se utilizan diferentes tipos de vectores virales, como los vectores adenovirales (AV) o los vectores virales adenoasociados (AAV). Estos Vectores Virales son cruciales para la entrega en el SNC.
Finalmente, existe un último esquema de terapia génica, en el que el vector viral se administra in situ, es decir, a un órgano o área específica del cuerpo del paciente, ya sea mediante inyección directa, por ejemplo, en un tumor (en el caso del melanoma) o en áreas cerebrales adecuadas (en el caso de neuropatías), o mediante la inserción de un catéter si el órgano a tratar es el corazón. La selección del procedimiento depende completamente del tipo de indicación, el tejido afectado y el tipo de célula que requiere corrección.
Desafíos y Consideraciones de Seguridad
El campo de la terapia génica ha enfrentado escepticismo y desafíos significativos a lo largo de los años. La trágica muerte de Jesse Gelsinger a principios de la década de 2000, probablemente debido a una respuesta inmune fatal desencadenada por vectores adenovirales, subrayó la falta de conocimiento sobre las interacciones vector-huésped y la necesidad de regulaciones más estrictas.
Uno de los principales riesgos de seguridad asociados con el uso de vectores virales, especialmente los retrovirales, es la mutagenicidad insercional o genotoxicidad. Esto implica la activación de protooncogenes o la interrupción de genes supresores de tumores debido a la integración del vector en el genoma del huésped. Aunque inicialmente se pensó que la integración sería aleatoria, se demostró que los vectores retrovirales tienen patrones de integración no aleatorios, lo que llevó a eventos de leucemia en ensayos clínicos para inmunodeficiencias (X-SCID) y otras enfermedades de la sangre. Esto impulsó la investigación para comprender los mecanismos de integración y desarrollar vectores más seguros, como los vectores lentivirales, que muestran patrones de integración diferentes, o los vectores autodestructivos (SIN) y los aisladores de cromatina, diseñados para minimizar el riesgo de activación de oncogenes vecinos.
Además de la mutagenicidad insercional, la respuesta inmune del huésped a los componentes del vector viral sigue siendo un desafío importante, particularmente con los AAV. Los anticuerpos neutralizantes preexistentes pueden reducir la eficacia de la transducción. Las cápsides exógenas de AAV pueden activar linfocitos T citotóxicos (CTL) que eliminan las células transducidas o generar anticuerpos neutralizantes, lo que impide la readministración. Ciertos serotipos de AAV pueden activar respuestas inmunes innatas a través de los receptores tipo Toll (TLR), produciendo citoquinas proinflamatorias. Si el transgén codifica una proteína extraña, también pueden generarse respuestas inmunes contra esta proteína. Se han reportado casos graves de hepatotoxicidad y patología del ganglio de la raíz dorsal asociados con la administración de AAV, especialmente a dosis altas.
Para las enfermedades neurológicas, la entrega segura, eficiente y selectiva de productos génicos en el SNC sigue siendo un desafío clave. La mayoría de los agentes administrados periféricamente no pueden cruzar la barrera hematoencefálica (BBB) o lo hacen de manera ineficiente. Esto a menudo requiere vías de administración invasivas como la intratecal o el uso de vectores virales (como AAVs diseñados) que puedan atravesar la BBB o ser administrados directamente en el SNC.
La Era de la Edición Genómica
En la última década, la Edición Genómica ha surgido como una poderosa evolución de la terapia génica. Tecnologías como las nucleasas de dedos de zinc (ZFNs), las nucleasas efectoras tipo activador de transcripción (TALENs) y, más prominentemente, el sistema `CRISPR/Cas9`, permiten modificaciones genómicas sitio-específicas, desde ediciones de un solo nucleótido hasta grandes deleciones o la integración dirigida de genes completos. A diferencia de la adición génica tradicional con vectores retrovirales, que pueden integrar el gen en lugares aleatorios, la edición genómica apunta a sitios específicos, lo que se espera que reduzca el riesgo de mutagenicidad insercional y permita mantener la regulación natural del gen.
El mecanismo principal de estas nucleasas implica inducir una ruptura de doble cadena (DSB) en la molécula de ADN, seguida de una reparación a través de la unión de extremos no homólogos (NHEJ) o la recombinación homóloga (HR). NHEJ puede introducir pequeñas inserciones o deleciones (indels) que interrumpen la secuencia objetivo, mientras que HR, con una plantilla donante, puede corregir la secuencia o insertar nuevo ADN en el sitio de la ruptura. Aunque las DSB ocurren naturalmente, las nucleasas diseñadas aumentan drásticamente la frecuencia de reparación dirigida.
La edición genómica presenta ventajas significativas, como la capacidad de corregir mutaciones "gain-of-function" o defectos en genes grandes, y la posibilidad de mantener la expresión fisiológica del gen. Sin embargo, también enfrenta desafíos. El más importante en términos de seguridad es la identificación de los efectos fuera de objetivo (off-target), donde la nucleasa corta o edita secuencias no deseadas en el genoma. Se han desarrollado métodos sofisticados para detectar estos efectos, pero su interpretación y relevancia clínica aún son complejas.
Otros desafíos incluyen la eficiencia de la edición en tipos celulares difíciles de modificar, la viabilidad celular después de la manipulación (especialmente con rupturas de doble cadena que activan la respuesta de daño al ADN), y la posibilidad de grandes reordenamientos cromosómicos. Se investigan estrategias para mejorar la eficiencia y seguridad, como modular las vías de reparación del ADN, silenciar temporalmente p53 para mejorar la viabilidad celular, o utilizar nanopartículas y enfoques como la edición de bases (base editing) o la edición de cebado (prime editing) que no requieren DSBs o son más precisos.
Un concepto importante en la edición genómica es la identificación y utilización de 'puertos seguros' genómicos (safe harbors): loci en el genoma que pueden aceptar la integración de un transgén o ser modificados sin causar efectos adversos significativos en genes vecinos o elementos reguladores esenciales. La integración dirigida en estos sitios ofrece una mayor seguridad y predictibilidad de la expresión del transgén.
Ejemplos de aplicación de la edición genómica incluyen la corrección de mutaciones específicas en enfermedades como la beta-talasemia o la enfermedad de células falciformes, o la inducción de hemoglobina fetal (HbF) mediante la interrupción de represores de la HbF, imitando un fenotipo de persistencia hereditaria de hemoglobina fetal (HPFH). Estos enfoques se están investigando activamente en ensayos clínicos.
Manufactura y Regulación
La transición de estas terapias de la investigación a la clínica y, finalmente, a la comercialización, implica complejidades considerables en la manufactura y la regulación. Existen modelos de manufactura centralizados, donde las células se recolectan localmente, se envían a una instalación central para la modificación genética y luego se devuelven al hospital. Este modelo es más familiar para las agencias reguladoras pero presenta desafíos logísticos y de tiempo, especialmente para productos con vida útil limitada.
Alternativamente, los modelos de manufactura descentralizados, más cercanos a la práctica de trasplante de células madre, implican la recolección y procesamiento local de las células. Esto ofrece mayor flexibilidad y personalización, pero requiere una estandarización y robustez excepcionales en múltiples sitios. La variabilidad del material de partida (células del paciente), las condiciones de cultivo y los procesos de manipulación son desafíos que deben abordarse, idealmente mediante la automatización y el uso temprano de materiales y procesos de grado GMP (Buenas Prácticas de Manufactura).
Las agencias reguladoras como la Agencia Europea de Medicamentos (EMA) y la Administración de Alimentos y Medicamentos (FDA) evalúan rigurosamente la seguridad y eficacia de estos productos, que a menudo son considerados 'fármacos vivos'. La definición de 'innovación' en este campo también es compleja y varía entre países, pero idealmente debe combinar seguridad, beneficio clínico significativo y asequibilidad para los sistemas de salud. El alto costo de algunas de estas terapias es un tema de discusión importante, a menudo denominado 'toxicidad financiera'.
Perspectivas Futuras
El campo de la terapia génica y la edición genómica para trastornos neurológicos está en rápida evolución. Los avances científicos han superado muchos de los obstáculos iniciales, llevando a terapias aprobadas y a un gran número de ensayos clínicos en curso para diversas afecciones neurodegenerativas y monogénicas.
Para desbloquear el verdadero potencial de estas terapias y lograr una aplicabilidad más amplia, es esencial abordar los desafíos restantes en seguridad (especialmente off-targets y efectos a largo plazo), optimizar los métodos de entrega al SNC, mejorar la eficiencia de la edición genómica, refinar los procesos de manufactura para garantizar la robustez y la asequibilidad, y navegar el complejo panorama regulatorio y de reembolso. La colaboración entre investigadores, médicos, industria, agencias reguladoras y responsables políticos será fundamental para el éxito futuro.
Aunque el campo aún se considera relativamente inmaduro, el progreso logrado en las últimas dos décadas, ejemplificado por el impacto en enfermedades como la SMA, subraya el enorme potencial de la terapia génica y la edición genómica para transformar el tratamiento de trastornos neurológicos previamente incurables, ofreciendo la esperanza de una corrección duradera en la raíz de la enfermedad.
Preguntas Frecuentes sobre Terapia Génica Neurológica
¿Qué es la Atrofia Muscular Espinal (SMA)?
Es una enfermedad neuromuscular genética causada por mutaciones en el gen SMN1, que provoca la pérdida de neuronas motoras y debilidad muscular progresiva.
¿Cómo funcionan las terapias génicas aprobadas para SMA?
Nusinersen (un ASO) modifica el empalme del gen SMN2 para producir más proteína SMN completa. Onasemnogene abeparvovec (un vector AAV9) entrega una copia funcional del gen SMN1 a las células.
¿Cuáles son los principales tipos de terapia génica?
Los tipos principales son ex vivo (modificar células fuera del cuerpo y reinfundirlas), in vivo (administrar el vector directamente en el cuerpo, como en el torrente sanguíneo o LCR) e in situ (inyección directa en un tejido u órgano específico).
¿Cuáles son los mayores desafíos de la terapia génica para el cerebro?
Los desafíos incluyen cruzar la barrera hematoencefálica, garantizar una entrega eficiente y extendida en el SNC, manejar las respuestas inmunes al vector y abordar los riesgos de seguridad como los efectos fuera de objetivo o la mutagenicidad insercional.
¿Qué es la edición genómica (CRISPR)?
Es una tecnología que permite modificar el ADN de manera precisa en sitios específicos del genoma, con el objetivo de corregir mutaciones o introducir cambios terapéuticos.
¿Es permanente la terapia génica?
Algunas terapias génicas, especialmente aquellas que utilizan vectores que se integran en el genoma o que se aplican a células no divisibles como las neuronas (con vectores no integradores como AAV), pueden ofrecer expresión génica a largo plazo. La edición genómica que corrige directamente el ADN es potencialmente permanente. Sin embargo, la necesidad de readministración o la persistencia varían según el tipo de terapia, el vector y las células objetivo.
Si quieres conocer otros artículos parecidos a Terapia Génica para Trastornos Neurológicos puedes visitar la categoría Neurociencia.