La Enfermedad de Huntington es una afección neurológica progresiva que, aunque poco común, ejerce un impacto devastador en quienes la padecen. Se caracteriza por el deterioro gradual de las células nerviosas en áreas clave del cerebro, lo que resulta en una compleja gama de síntomas que afectan el movimiento, la capacidad cognitiva y la salud mental. Esta enfermedad, a menudo hereditaria, presenta un desafío tanto para los pacientes como para sus familias, alterando profundamente la calidad de vida a medida que avanza.

A diferencia de otras enfermedades neurodegenerativas, la de Huntington suele manifestarse en la edad adulta, típicamente entre los 30 y 40 años, aunque existe una forma juvenil de inicio más temprano y rápida progresión. Comprender qué sucede en el cerebro a nivel celular y genético es fundamental para abordar sus complejas manifestaciones.
- Los Síntomas: Un Espectro Complejo de Afecciones
- El Cerebro Bajo Ataque: Neurodegeneración en Huntington
- Causas y Factores de Riesgo: La Herencia Genética
- Complicaciones y Pronóstico
- Prevención y Planificación Familiar
- Tabla Comparativa: Síntomas Típicos (Adultos vs. Juvenil)
- Preguntas Frecuentes sobre la Enfermedad de Huntington
Los Síntomas: Un Espectro Complejo de Afecciones
La Enfermedad de Huntington no se limita a un solo tipo de síntoma; en cambio, presenta un amplio espectro que varía enormemente de una persona a otra y cambia a lo largo del curso de la enfermedad. Estos síntomas pueden manifestarse en tres dominios principales: trastornos del movimiento, afecciones cognitivas y problemas de salud mental.
Trastornos del Movimiento
Uno de los sellos distintivos de la Enfermedad de Huntington es la presencia de movimientos involuntarios. El más conocido es la corea, que se manifiesta como movimientos espasmódicos o de contorsión incontrolables que pueden afectar cualquier parte del cuerpo, especialmente brazos, piernas, cara y lengua. Sin embargo, la enfermedad también impacta la capacidad para realizar movimientos voluntarios, lo cual puede ser incluso más incapacitante que la corea, dificultando tareas cotidianas como caminar, mantener el equilibrio, hablar o tragar.
- Movimientos espasmódicos o de contorsión involuntarios (corea)
- Rigidez o contracturas musculares
- Movimientos oculares lentos o inusuales
- Dificultad para caminar, mantener la postura y el equilibrio
- Problemas para hablar o tragar
La pérdida del control sobre los movimientos voluntarios afecta directamente la independencia del individuo, su capacidad para trabajar y comunicarse.
Alteraciones Cognitivas
La enfermedad también deteriora las capacidades cognitivas, afectando la forma en que una persona piensa, razona y resuelve problemas. Estos síntomas a menudo incluyen:
- Dificultad para organizar, establecer prioridades o concentrarse
- Perseveración: quedarse 'atascado' en un pensamiento o acción
- Falta de control de impulsos, que puede llevar a arrebatos o conductas imprudentes
- Falta de conciencia sobre las propias limitaciones o comportamientos
- Lentitud para procesar información o encontrar las palabras adecuadas
- Problemas para aprender información nueva
Estas dificultades cognitivas impactan la capacidad de planificación, la toma de decisiones y la interacción social, contribuyendo al aislamiento y la frustración.
Impacto en la Salud Mental
Los problemas de salud mental son una parte integral de la Enfermedad de Huntington, a menudo manifestándose antes o al mismo tiempo que los síntomas motores y cognitivos. La depresión es la afección mental más común, y no es simplemente una reacción al diagnóstico, sino que parece estar directamente relacionada con los cambios y daños en el cerebro. Los síntomas depresivos pueden incluir irritabilidad, tristeza, apatía, aislamiento social, problemas de sueño, fatiga e incluso pensamientos suicidas.
Otras afecciones mentales comunes asociadas incluyen el Trastorno Obsesivo-Compulsivo (TOC), manía (estado de ánimo elevado, hiperactividad) y trastorno bipolar (alternancia entre episodios de depresión y manía). La pérdida de peso inexplicable también es frecuente a medida que la enfermedad progresa.
Síntomas en la Enfermedad de Huntington Juvenil
Cuando la enfermedad se inicia antes de los 20 años, presenta algunas diferencias:
- Cambios conductuales: problemas de atención, bajo rendimiento escolar, agresividad.
- Cambios físicos: músculos rígidos (afectando la marcha), temblores leves, caídas frecuentes, convulsiones.
La progresión en la forma juvenil suele ser más rápida.
El Cerebro Bajo Ataque: Neurodegeneración en Huntington
La esencia de la Enfermedad de Huntington reside en el daño progresivo a células nerviosas específicas en el cerebro. La investigación ha identificado áreas particularmente vulnerables y procesos moleculares involucrados.
El Rol Clave de los Ganglios Basales
La enfermedad afecta de manera prominente los ganglios basales, un grupo de estructuras cerebrales profundas cruciales para el control del movimiento, el comportamiento y la cognición. Dentro de los ganglios basales, el estriado (formado por el núcleo caudado y el putamen) es una de las regiones más afectadas en las etapas tempranas de la enfermedad.
La disfunción y pérdida de neuronas en el estriado, particularmente de un tipo llamado neuronas espinosas medianas (MSNs), alteran los complejos circuitos que regulan el movimiento, explicando la corea y las dificultades con los movimientos voluntarios. Se ha observado una pérdida diferencial de ciertas fibras neuronales dentro de los ganglios basales, como la pérdida temprana y progresiva de fibras ENK+ en el Globo Pálido externo (GPe), mientras que las fibras SP+ en el Globo Pálido interno (GPi) se ven afectadas más tardíamente.
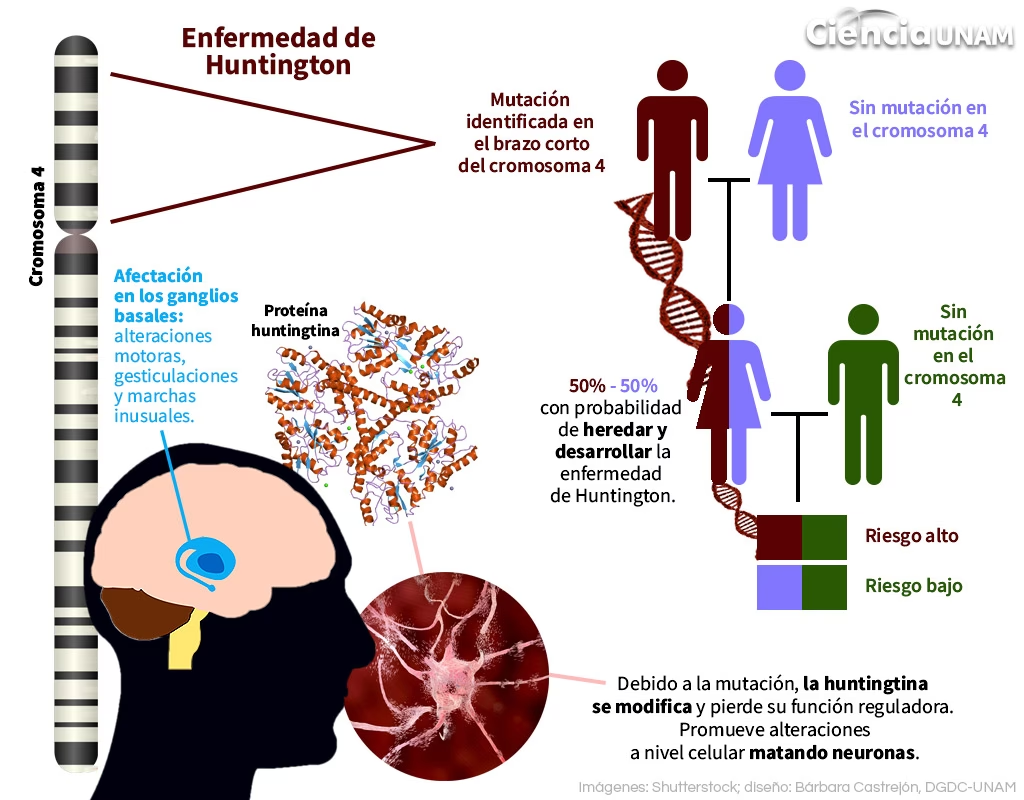
La Proteína Huntingtina y la Mutación Genética
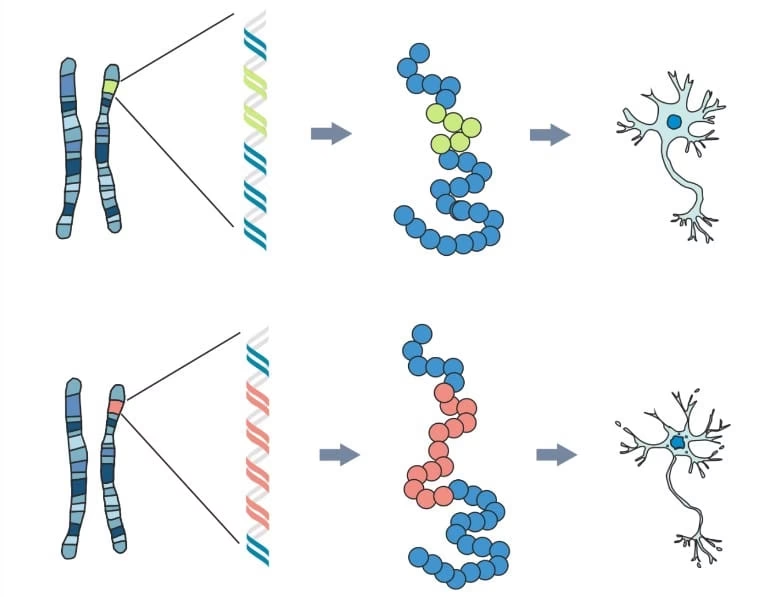
La causa principal de la Enfermedad de Huntington es una mutación en un único gen, conocido como gen IT15 o HTT, que codifica la proteína huntingtina. Esta mutación consiste en una expansión anormal de una secuencia repetida de ADN (CAG) dentro del gen. Normalmente, esta repetición se encuentra entre 9 y 35 veces. Sin embargo, en las personas con Huntington, la repetición de CAG es superior a 35.
Esta expansión de CAG se traduce en una proteína huntingtina anormalmente larga, con una cadena extendida de glutaminas (un aminoácido) en su extremo N-terminal. Aunque la proteína huntingtina normal tiene funciones importantes en el cerebro, la forma mutada se vuelve tóxica, aunque el mecanismo exacto de esta toxicidad aún se investiga activamente. Se cree que la enfermedad es principalmente un trastorno de 'ganancia de función', donde la proteína mutada adquiere propiedades dañinas nuevas o exageradas.
La longitud de la repetición de CAG se correlaciona con la gravedad de la enfermedad y la edad de inicio: a mayor número de repeticiones, más temprana y severa tiende a ser la enfermedad. Las repeticiones entre 36 y 39 pueden tener una penetrancia reducida, lo que significa que no todas las personas con este rango desarrollarán síntomas durante su vida. La estabilidad de la repetición de CAG puede verse influenciada por factores genéticos, y se ha observado que las expansiones son más frecuentes y significativas en la transmisión paterna.
Neuroinflamación y Neurogénesis
El cerebro afectado por Huntington muestra signos de neuroinflamación, caracterizada por la activación de células inmunes residentes llamadas microglía. Esta activación microglial es una característica patológica prominente que se observa desde etapas tempranas de la enfermedad. La microglía activada libera mediadores inflamatorios que, si bien inicialmente podrían intentar proteger el tejido, su activación sostenida puede exacerbar el daño neuronal.
Curiosamente, en respuesta a la lesión en el estriado, se observa un aumento en la producción de células progenitoras en la zona subventricular (SVZ) del cerebro. Estas células migran hacia las áreas dañadas, diferenciándose en neuronas y células gliales. Este proceso de neurogénesis aumentada parece ser un intento del cerebro por reparar el daño, aunque no es suficiente para detener la progresión de la enfermedad.
Causas y Factores de Riesgo: La Herencia Genética
La causa principal y casi única de la Enfermedad de Huntington es la herencia de la mutación en el gen HTT. La enfermedad sigue un patrón de herencia autosómico dominante. Esto significa que una persona solo necesita heredar una copia mutada del gen de uno de sus padres para desarrollar la enfermedad.
Cada hijo de un progenitor afectado tiene un 50% de probabilidad de heredar el gen mutado y, por lo tanto, de desarrollar la enfermedad. Este patrón de herencia es un factor de riesgo clave. Las personas con antecedentes familiares conocidos de Huntington tienen un riesgo significativo y pueden considerar pruebas genéticas para determinar si portan la mutación.
Complicaciones y Pronóstico
Una vez que comienzan los síntomas, la capacidad funcional de una persona con Enfermedad de Huntington disminuye progresivamente. La velocidad de progresión varía, pero generalmente, desde la aparición de los primeros síntomas hasta el fallecimiento, transcurren entre 10 y 30 años en adultos, y entre 10 y 15 años en la forma juvenil.
Las complicaciones son numerosas y reflejan el deterioro general. El riesgo de suicidio aumenta, particularmente antes del diagnóstico y a medida que la persona pierde independencia. En las etapas avanzadas, los pacientes requieren asistencia total para todas las actividades diarias, quedan postrados en cama y, a menudo, pierden la capacidad de hablar, aunque generalmente conservan la comprensión del lenguaje y el reconocimiento de seres queridos.
Las causas comunes de fallecimiento en personas con Enfermedad de Huntington incluyen:
- Neumonía u otras infecciones (a menudo relacionadas con la dificultad para tragar o la inmovilidad)
- Lesiones por caídas
- Complicaciones derivadas de los problemas para tragar (como asfixia o desnutrición)
Prevención y Planificación Familiar
Dado su carácter genético, la prevención de la Enfermedad de Huntington se centra en la planificación familiar y las pruebas genéticas para personas en riesgo. Aquellos con antecedentes familiares pueden buscar asesoramiento genético para entender su riesgo y las opciones disponibles.
Las opciones para parejas en riesgo que desean tener hijos incluyen:
- Pruebas prenatales: Realizar pruebas al feto durante el embarazo para detectar la presencia del gen mutado.
- Fecundación In Vitro (FIV) con donación de gametos: Utilizar esperma u óvulos de un donante que no porte la mutación.
- FIV con Diagnóstico Genético Preimplantacional (DGP): En este proceso, los embriones creados en laboratorio mediante FIV son analizados genéticamente antes de ser implantados en el útero. Solo se seleccionan e implantan los embriones que no portan el gen de Huntington.
Estas opciones permiten a las parejas tomar decisiones informadas para evitar transmitir la enfermedad a sus hijos.
Tabla Comparativa: Síntomas Típicos (Adultos vs. Juvenil)
| Síntoma | Enfermedad de Huntington (Adultos) | Enfermedad de Huntington Juvenil (<20 años) |
|---|---|---|
| Inicio | Generalmente 30-40 años | Antes de los 20 años |
| Progresión | Típicamente más lenta (10-30 años hasta el fallecimiento) | Generalmente más rápida (10-15 años hasta el fallecimiento) |
| Movimientos Involuntarios (Corea) | Prominente en etapas tempranas y medias | Puede ser menos prominente inicialmente; rigidez y lentitud más comunes al principio |
| Rigidez Muscular / Distonía | Puede aparecer, a menudo en etapas posteriores | Frecuente y significativa desde el inicio, afectando la marcha |
| Temblor | Menos común | Puede estar presente al principio |
| Caídas / Torpeza | Común a medida que avanza la enfermedad | Frecuente desde etapas tempranas |
| Convulsiones | Poco común | Más frecuente |
| Cambios Conductuales | Irritabilidad, apatía, aislamiento, impulsividad | Problemas de atención, rendimiento escolar bajo, agresividad |
| Problemas Cognitivos | Dificultad de organización, perseveración, lentitud de pensamiento | Similares a los adultos, pero el impacto en el aprendizaje es crucial |
| Problemas de Salud Mental | Depresión, TOC, Manía, Bipolaridad (comunes) | Similares, pero la irritabilidad y agresividad pueden ser más destacadas |
| Pérdida de Peso | Común a medida que avanza | Común |
Preguntas Frecuentes sobre la Enfermedad de Huntington
- ¿La Enfermedad de Huntington es curable?
- Actualmente, no existe cura para la Enfermedad de Huntington. Los tratamientos disponibles se centran en manejar los síntomas para mejorar la calidad de vida, pero no pueden detener la progresión del daño cerebral.
- ¿Cómo se diagnostica la Enfermedad de Huntington?
- El diagnóstico se basa en la evaluación de los síntomas neurológicos (motores, cognitivos, psiquiátricos) y un historial familiar positivo. Se confirma mediante una prueba genética que detecta la expansión anormal de la repetición de CAG en el gen HTT.
- Si un padre tiene el gen, ¿significa que su hijo definitivamente tendrá la enfermedad?
- No definitivamente, pero hay un alto riesgo. Debido a que sigue un patrón de herencia autosómico dominante, cada hijo de un progenitor afectado tiene un 50% de probabilidad de heredar la copia mutada del gen. Si heredan el gen mutado (con repeticiones >35), desarrollarán la enfermedad en algún momento de su vida, aunque la edad de inicio puede variar.
- ¿Qué es exactamente la corea?
- La corea es un tipo de trastorno del movimiento caracterizado por movimientos involuntarios, rápidos, espasmódicos y que parecen danzar. Afecta diferentes partes del cuerpo y es uno de los síntomas motores más reconocidos de la Enfermedad de Huntington, aunque no es el único.
- ¿La pérdida de memoria es un síntoma principal?
- Si bien los problemas cognitivos son significativos en la Enfermedad de Huntington, que incluyen dificultades con el procesamiento de información y el aprendizaje nuevo, la pérdida de memoria severa en el sentido de amnesia profunda no es típicamente el síntoma cognitivo inicial o principal, como puede ser en otras demencias. Las dificultades se centran más en la función ejecutiva (planificación, organización) y la velocidad del pensamiento, aunque la memoria a largo plazo puede verse afectada en etapas avanzadas. El texto provisto menciona 'se suele perder la memoria' en el transcurso de los años, indicando que es un síntoma que aparece con la progresión.
La Enfermedad de Huntington es una compleja afección neurodegenerativa con profundas raíces genéticas que afecta de manera selectiva áreas cerebrales clave como los ganglios basales. El entendimiento de la mutación en la proteína huntingtina, los mecanismos de toxicidad neuronal, la respuesta inflamatoria y los intentos de reparación del cerebro continúa expandiéndose gracias a la investigación científica. Aunque actualmente no hay cura, el manejo sintomático y las opciones de planificación familiar ofrecen herramientas importantes para las personas afectadas y sus familias, mientras la comunidad científica trabaja incansablemente en busca de terapias que puedan detener o revertir su devastador curso.
Si quieres conocer otros artículos parecidos a ¿Qué Pasa en el Cerebro con Huntington? puedes visitar la categoría Neurociencia.