La minería de datos, en el ámbito de la neurociencia, representa un enfoque computacional innovador que permite analizar una gran cantidad de conjuntos de datos disponibles públicamente. Su objetivo principal es generar nuevas hipótesis que puedan ser validadas experimentalmente, mejorando así nuestra comprensión de la patogénesis de las enfermedades neurodegenerativas. A medida que el número de conjuntos de datos de secuenciación aumenta, el análisis de microarrays, realizado en diversas muestras biológicas, sigue constituyendo una vasta colección de datos con múltiples programas web que facilitan un análisis eficiente y conveniente.
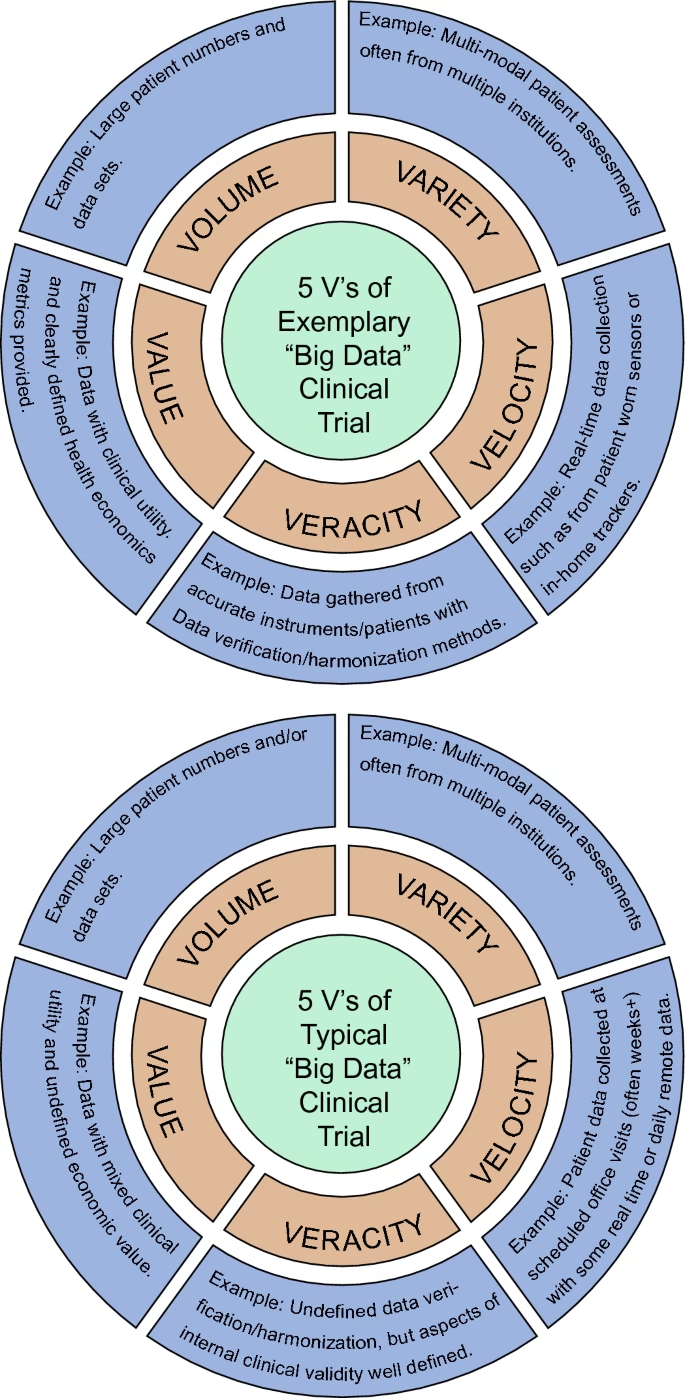
Este campo emergente se centra en extraer conocimiento valioso de la complejidad de los datos biológicos, permitiendo a los investigadores identificar patrones ocultos, correlaciones y tendencias que de otra manera serían difíciles de detectar. Al integrar información de diversas fuentes y experimentos, la minería de datos ofrece una visión más holística de los sistemas biológicos afectados por trastornos neurológicos. Desde el descubrimiento de biomarcadores hasta el desarrollo de nuevas estrategias terapéuticas, su potencial para transformar la neurociencia traslacional es inmenso.
- Muestras Biológicas Clave para el Análisis de Datos
- Análisis de Datos Transcriptómicos: Microarrays vs. RNA-Seq
- La Tubería del Análisis de Datos de Microarrays
- Aplicaciones de la Minería de Datos en Neurociencia Traslacional
- Salvando Brechas y Perspectivas Futuras
- Preguntas Frecuentes sobre Minería de Datos en Neurociencia
Muestras Biológicas Clave para el Análisis de Datos
La selección adecuada de muestras biológicas es fundamental en la minería de datos para la neurociencia, ya que determina los resultados y conclusiones de los estudios. En este campo, las muestras más comunes incluyen tejidos cerebrales post-mortem, líquido cefalorraquídeo (LCR), sangre periférica y células madre humanas. Estas muestras, recolectadas de controles sanos o pacientes con enfermedades neurodegenerativas, se someten a diversas mediciones biológicas cuantitativas y cualitativas, como caracterizaciones de microarrays y análisis de imágenes, para comprender la funcionalidad cerebral en diversas etapas de la enfermedad.
Es crucial clasificar las muestras biológicas basándose en información demográfica básica, genotipos (por ejemplo, pacientes con mutaciones patogénicas), fenotipos (características observables de la enfermedad) y resultados clínicos (como la etapa de Braak en AD). Otro factor importante es la facilidad de obtención de las muestras, considerando los procedimientos experimentales y su carácter invasivo. La aplicación del estudio (descubrimiento de biomarcadores, desarrollo de fármacos, elucidación de mecanismos de enfermedad) también guía la selección de la muestra.
Tejidos Cerebrales Post-mortem
El uso de tejidos cerebrales humanos post-mortem proporciona observaciones directas sobre la patología y el estado de la enfermedad en el momento del fallecimiento del paciente. Sin embargo, su principal limitación es la imposibilidad de obtener muestras a lo largo del tiempo en pacientes vivos, lo que introduce un sesgo y dificulta la evaluación de las etapas iniciales de la enfermedad y la elucidación del curso de la progresión. La heterogeneidad del tejido, incluyendo diversidad, variabilidad y baja reproducibilidad entre sujetos, es otro desafío significativo. A pesar de estas dificultades, el análisis combinado de múltiples conjuntos de datos de tejidos cerebrales, con un gran número de pacientes, promete un análisis más preciso para el descubrimiento de biomarcadores y una mejor comprensión de los mecanismos de enfermedades neurológicas.
Líquido Cefalorraquídeo (LCR) y Sangre Periférica
Para perfilar pacientes vivos, el LCR y la sangre periférica son muestras biológicas frecuentemente utilizadas debido a su facilidad de extracción y sus aplicaciones diagnósticas, y son menos propensas a la heterogeneidad. Mientras que la sangre periférica tiene amplias aplicaciones, la aplicación principal del LCR es la detección y diagnóstico de enfermedades neurológicas. Biomarcadores establecidos en LCR, como β-amiloide y tau, son útiles para el diagnóstico temprano de AD, mientras que la α-sinucleína y las cadenas ligeras de neurofilamentos ayudan en el diagnóstico de PD y MS, respectivamente.
El establecimiento de biomarcadores en sangre ha sido una estrategia muy buscada debido a su mínima invasividad, bajo costo y accesibilidad. Aunque se trabaja para aumentar la precisión y correlación de los biomarcadores sanguíneos con los del LCR, la necesidad de más estudios de correlación y nuevos métodos de análisis que consideren la variabilidad interindividual es apremiante. Estudios recientes se centran en los niveles de expresión de miRNA en sangre periférica por sus características de biomarcador, como abundancia, especificidad tisular y estabilidad. Actualmente, LCR y sangre se usan a menudo en combinación para evaluar la enfermedad, validando el estado de la enfermedad con biomarcadores de LCR y aislando genes diferencialmente expresados (DEGs) de sangre periférica para reforzar la credibilidad de los biomarcadores sanguíneos propuestos.
Células Madre Humanas
El uso de células madre humanas está en aumento, especialmente las células madre pluripotentes inducidas (iPSCs) derivadas de pacientes. Estas células son modelos importantes para entender los mecanismos de enfermedad y probar cómo los fármacos alteran los perfiles de ARN, la expresión de proteínas y las funciones celulares. Las iPSCs pueden diferenciarse en diversos tipos celulares del sistema nervioso central (SNC) y contribuir a la generación de organoides multicelulares. Sus ventajas incluyen la proliferación con potencial de desarrollo, facilidad de modificación genética y modelado directo de la biología humana sin factores de confusión específicos de especie. El análisis de datos de iPSCs es prometedor para el descubrimiento de biomarcadores y el desarrollo terapéutico.
Análisis de Datos Transcriptómicos: Microarrays vs. RNA-Seq
Las tecnologías para cuantificar el transcriptoma han evolucionado significativamente, desde los microarrays y la PCR cuantitativa hasta el RNA-seq masivo y el RNA-seq de célula única (sc/snRNA-seq). Aunque el RNA-seq está a la vanguardia para comprender la heterogeneidad de enfermedades neurológicas como AD, PD y MS, y detectar secuencias y variantes de empalme novedosas, presenta desafíos en el análisis de datos, almacenamiento y posible sesgo de longitud de transcripción.
Los microarrays, aunque limitados a transcripciones establecidas, son capaces de detectar genes altamente variables. A pesar de las diferencias técnicas, los resultados de microarray y RNA-seq muestran una alta consistencia. En el contexto de la Minería de Datos, los microarrays siguen siendo ampliamente adoptados debido a su bajo costo, alta eficiencia, sesgo limitado, mayor potencia estadística y la gran cantidad de conjuntos de datos públicos disponibles en neurociencia. Ambas técnicas son complementarias y la integración de datos de ambas puede proporcionar análisis más completos.
La Tubería del Análisis de Datos de Microarrays
El método de microarray ha sido uno de los más comunes para el análisis transcriptómico, utilizado para identificar transcripciones codificantes de proteínas o ARN no codificantes diferencialmente expresados en estados de enfermedad. Existen múltiples bases de datos que archivan conjuntos de datos de microarrays, siendo la base de datos Gene Expression Omnibus (GEO) la predominante.
El proceso de minería de datos de microarrays generalmente sigue una tubería (pipeline) definida:
- Identificación de Conjuntos de Datos: Buscar conjuntos de datos adecuados en bases de datos públicas como GEO. Herramientas como GEO2R facilitan la comparación de grupos de muestras.
- Control de Calidad y Preprocesamiento: Normalizar los datos brutos, evaluar la calidad de la alineación y considerar posibles especies contaminantes. La selección de características puede ser necesaria para eliminar genes con expresión constante.
- Análisis Estadístico: Identificar genes diferencialmente expresados (DEGs) con significancia estadística (valores P) y magnitud del cambio (LogFC). Herramientas como el paquete limma en R, utilizado por GEO2R, son comunes. Se pueden generar gráficos como volcán y box plots, o UMAP.
- Análisis Funcional y Anotación: Elucidar las anotaciones funcionales de los DEGs y su papel en los mecanismos de enfermedad. Se utilizan herramientas web y de aplicación para análisis de vías (DAVID, IPA, GSEA, etc.) y visualización de redes (STRING, Cytoscape, NetworkAnalyst). Estos análisis ayudan a identificar vías biológicas disfuncionales y genes centrales (hub genes) en el contexto de la enfermedad.
Es importante destacar que algunas herramientas, como GSEA, consideran todos los valores de expresión transcriptómica, no solo los DEGs aislados, permitiendo un análisis más holístico de la desregulación génica.
Aplicaciones de la Minería de Datos en Neurociencia Traslacional
La minería de datos en neurociencia ha generado conocimientos significativos con aplicaciones directas en el campo traslacional.
Descubrimiento de Biomarcadores
El análisis de datos ha sido fundamental para identificar posibles biomarcadores de enfermedades neurodegenerativas. Por ejemplo, estudios de minería de datos han identificado genes diferencialmente expresados en tejidos cerebrales y sangre de pacientes con AD y PD, y en LCR de pacientes con MS. Genes como GSN, BDNF, TIMP1, VLDLR y APLP2 han sido validados como posibles biomarcadores de AD tanto bioinformáticamente como experimentalmente en sangre periférica. En MS, genes como NLRP3, LILRB2, C1QB, CD86, C1QA, CSF1R, IL1B y TLR2 han sido identificados como desregulados en LCR mediante minería de datos.

Desarrollo Terapéutico y Elucidación de Mecanismos
La minería de datos ayuda a comprender los mecanismos patogénicos identificando vías biológicas disfuncionales y genes clave. Por ejemplo, se ha demostrado la desregulación de genes y vías en AD, PD y MS. La identificación de genes como TYROBP en AD, implicado en la fagocitosis por microglía, o SRRM2 en PD, un factor de empalme de ARN, ilustra cómo el análisis de datos revela actores clave en la patogénesis. El uso de iPSCs derivadas de pacientes en conjunto con la minería de datos permite probar el efecto de compuestos terapéuticos en modelos relevantes y obtener información sobre cómo alteran los perfiles de expresión génica.
Análisis de ARN No Codificante
Además del ARNm, la minería de datos se aplica al análisis de ARN no codificante (ncRNA) como miRNA, circRNA y lncRNA, que desempeñan roles cruciales en el desarrollo neural, procesos cognitivos y regulación génica. Estos ncRNAs son cada vez más investigados como biomarcadores y en la elucidación de mecanismos de enfermedad. Estudios han identificado miRNAs desregulados en sangre y tejidos cerebrales de pacientes con MS y AD. Por ejemplo, la desregulación de hsa-miR-328-3p, hsa-miR-20a-5p y miR-196 en sangre de pacientes con MS, o la desregulación de hsa-miR-186-5p, hsa-miR-125a-3p, hsa-miR-22-3p, hsa-miR-24-3p, hsa-miR-6131 y hsa-miR-125b-1-3p en sangre de pacientes con AD. circRNAs y lncRNAs también muestran desregulación en enfermedades neurodegenerativas, con ejemplos como los circRNAs vinculados a genes de patología de AD (DOCK1, NTRK2, DLG1, KIF1B, TRAPPC9, APC) o el lncRNA NEAT1 altamente desregulado en el córtex entorrinal de pacientes con AD, con potencial como biomarcador.
Salvando Brechas y Perspectivas Futuras
Aunque los microarrays y el RNA-seq se diferencian en detalles técnicos (hibridación vs. conteo de lecturas, detección de secuencias nuevas, capacidad unicelular), ambos proporcionan mediciones de expresión génica. Una brecha importante es la estandarización de las tuberías de análisis para procesar datos de diferentes métodos. Sin embargo, la creciente disponibilidad de herramientas web y basadas en aplicaciones (ver Tabla 2) está facilitando el análisis de datos de alto contenido para ambos tipos de tecnologías.
| Característica | Microarray | RNA-Seq |
|---|---|---|
| Costo por muestra | Generalmente menor | Generalmente mayor (especialmente sc/snRNA-seq) |
| Eficiencia | Mayor | Menor (análisis más complejo) |
| Detección de secuencias/isoformas nuevas | Limitado a transcripciones conocidas | Sí |
| Sesgo de longitud de transcripción | Limitado | Puede tener (mitigado por DRS) |
| Datos públicos disponibles en neurociencia | Vasto | Creciente |
| Análisis unicelular | Reportado pero menor resolución/heterogeneidad | Alta capacidad (sc/snRNA-seq) |
| Análisis de datos | Más directo, herramientas web estandarizadas (GEO2R) | Más complejo, requiere más conocimiento de programación, menos estandarizado |
| Almacenamiento de datos | Menor | Mayor |
| Potencia estadística | Mayor en algunos contextos | Depende del número de lecturas y diseño experimental |
La capacidad de comparar y conciliar los resultados de microarray y RNA-seq es vital. Estudios muestran una alta correlación entre los valores de expresión obtenidos por ambos métodos. Es crucial utilizar múltiples conjuntos de datos, métodos de análisis rigurosos y parámetros estadísticos estrictos para reducir falsos positivos, especialmente con tamaños de muestra pequeños.
El futuro de la minería de datos en neurociencia apunta hacia la integración multi-ómica, combinando datos transcriptómicos con genómicos, proteómicos, metabolómicos, etc., para obtener una comprensión aún más profunda de las enfermedades. Esta integración es clave para avanzar en el fenotipado de precisión y la medicina personalizada para las enfermedades neurodegenerativas. La combinación de diferentes tipos de conjuntos de datos, junto con la validación experimental, es necesaria para análisis más completos.
Preguntas Frecuentes sobre Minería de Datos en Neurociencia
Aquí respondemos algunas preguntas comunes sobre este tema:
- ¿Qué tipos de datos se utilizan en la minería de datos en neurociencia? Se utilizan principalmente datos de transcriptómica (microarrays, RNA-seq) de muestras biológicas como tejidos cerebrales, LCR, sangre periférica y células madre. También se pueden incluir datos genómicos, proteómicos, y de otras 'ómicas', así como datos de muestras como plasma, orina, heces, microbioma intestinal, etc.
- ¿Por qué es importante utilizar conjuntos de datos públicos? El uso de conjuntos de datos disponibles públicamente permite a los investigadores analizar un gran volumen de datos de diversas fuentes y experimentos, aumentando la potencia estadística, validando hallazgos y generando nuevas hipótesis sin la necesidad de generar todos los datos desde cero.
- ¿Cómo ayuda la minería de datos a entender las enfermedades neurodegenerativas? Ayuda a identificar genes y vías desreguladas, descubrir posibles biomarcadores para diagnóstico y pronóstico, y encontrar posibles dianas terapéuticas. Permite analizar la complejidad de la enfermedad desde una perspectiva de sistemas, integrando información de diferentes tipos de muestras y tecnologías.
- ¿La minería de datos en neurociencia se limita solo al análisis de genes? No, aunque el análisis transcriptómico (ARNm y ARN no codificante como miRNA, circRNA, lncRNA) es muy común, también se aplica a datos de otras 'ómicas' como proteómica y metabolómica, así como a datos de imágenes y datos clínicos para obtener una visión más completa de la enfermedad.
- ¿Qué herramientas se utilizan para la minería de datos transcriptómicos? Existen diversas herramientas tanto web como de aplicación, como GEO2R, BART, DAVID, STRING, GSEA para microarrays, y BEAVR, RNAlysis, ScAmpi, ASAP para RNA-seq, entre otras.
En conclusión, la minería de datos es una herramienta poderosa y esencial en la neurociencia moderna. Al aprovechar la riqueza de los datos biológicos disponibles, particularmente los transcriptómicos de diversas muestras, los investigadores están haciendo progresos significativos en la comprensión de las complejas bases moleculares de las enfermedades neurodegenerativas. La continua mejora de las tecnologías y las herramientas de análisis, junto con la integración de datos multi-ómicos, allana el camino para diagnósticos más tempranos, terapias más efectivas y, en última instancia, una medicina de precisión para los trastornos del cerebro.
Si quieres conocer otros artículos parecidos a Minería de Datos en Neurociencia Explicada puedes visitar la categoría Neurociencia.