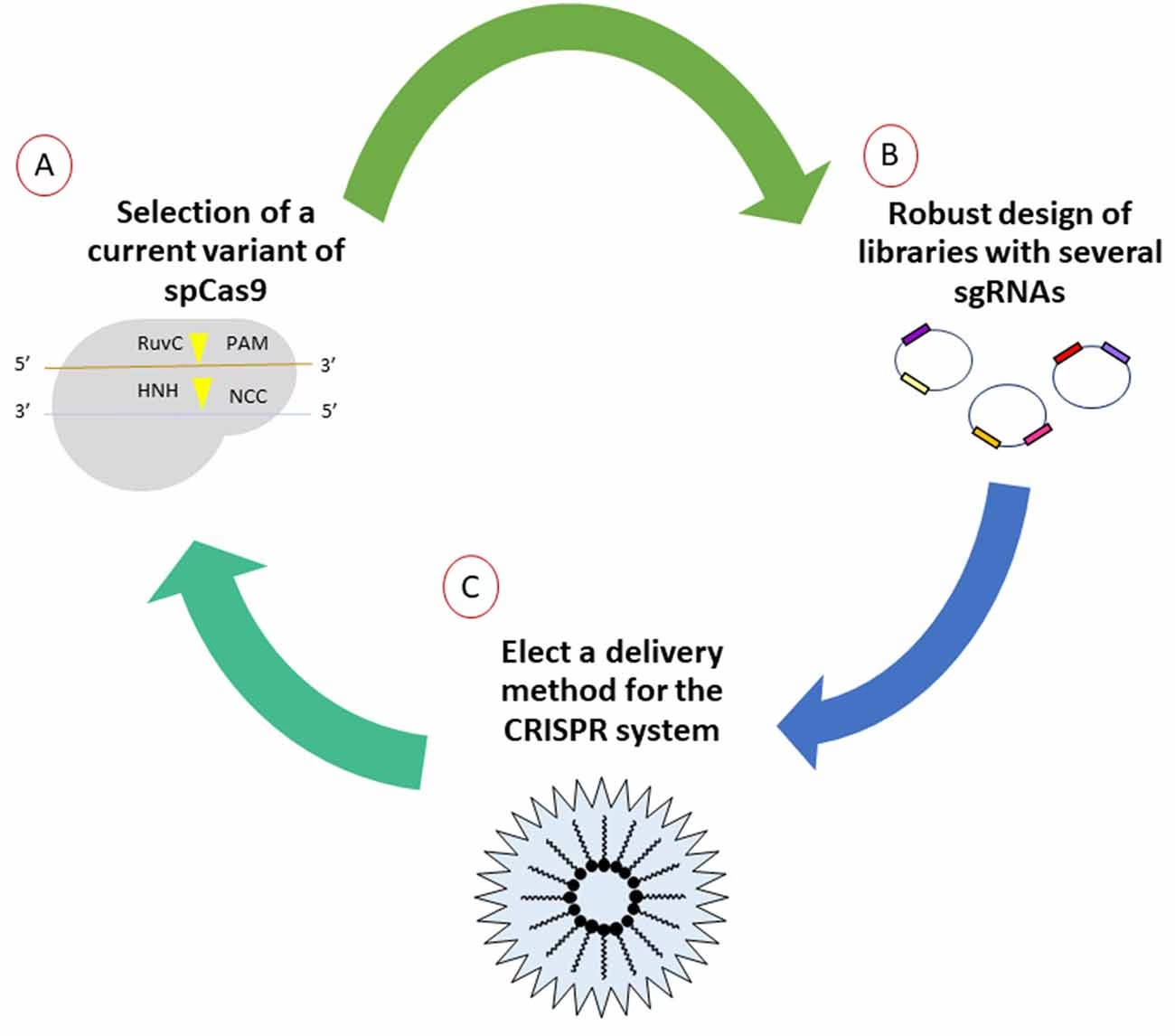
La edición del genoma basada en CRISPR-Cas9 representa una herramienta poderosa, altamente precisa, programable y accesible para investigar y manipular la expresión génica. Esta tecnología se deriva del sistema inmunitario adaptativo presente en arqueas y eubacterias, que les permite defenderse de ácidos nucleicos extraños como plásmidos y bacteriófagos. CRISPR significa 'Clustered Regularly Interspaced Short Palindromic Repeats' (Repeticiones Palindrómicas Cortas Agrupadas y Espaciadas Regularmente), y Cas9 (CRISPR associated protein-9) es una enzima endonucleasa que corta el ADN en sitios diana específicos. Esta inmunidad es heredable, lo que lleva a la formación de nuevas colonias resistentes a virus previamente encontrados.

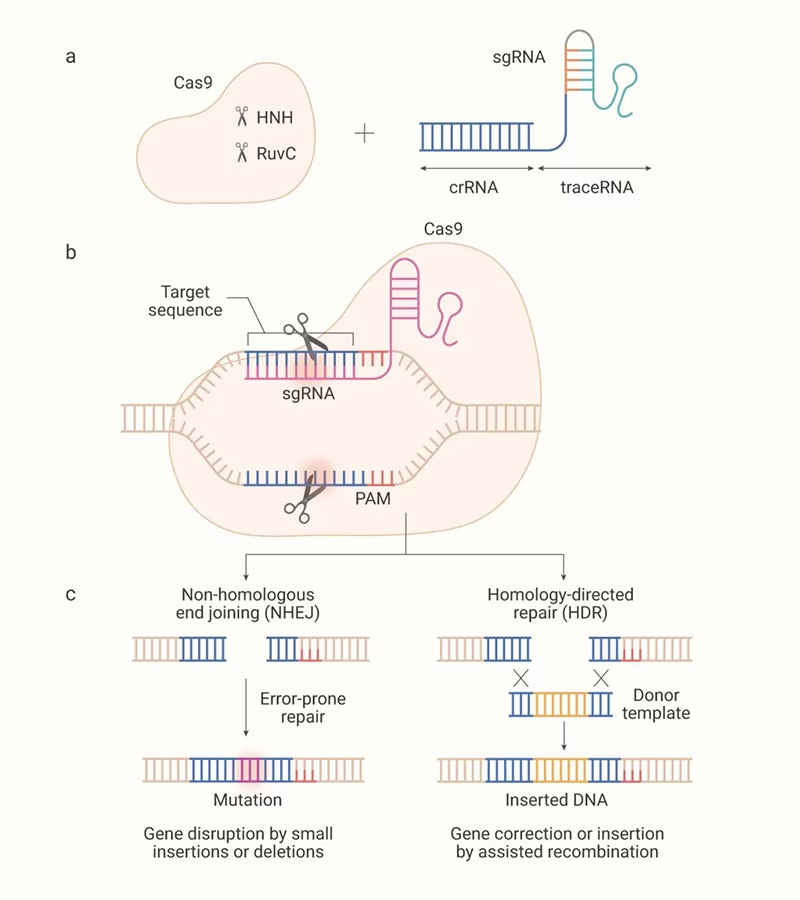
El complejo CRISPR-Cas9 está compuesto por un ARN guía simple (sgRNA o gRNA) altamente personalizable, que se une a la nucleasa Cas9. El sgRNA tiene dos componentes: una secuencia personalizable de aproximadamente 20 nucleótidos llamada 'espaciador' y una secuencia de andamiaje que se une y mantiene a la enzima Cas9. La Cas9 reconoce un Motivo Adyacente a Protoespaciador (PAM), que es una secuencia 5′-NGG o 5′-NAG (donde N es cualquier nucleótido) ubicada en el sitio diana. La enzima Cas9 posee dos dominios nucleasa, RuvC y HNH, que cortan diferentes hebras del ADN, generando roturas de doble cadena (DSBs). Estas DSBs son reparadas por la maquinaria celular de reparación del ADN, lo que conduce a cambios (deseados o no) en la secuencia.
La tecnología basada en CRISPR es más eficiente y precisa que otros sistemas de edición genética conocidos como TALENs (nucleasas efectoras tipo activador de transcripción) y ZFNs (nucleasas de dedo de zinc). Estos métodos anteriores son costosos, presentan mayores efectos fuera del objetivo (off-target) y toxicidad celular. La entrega exitosa de estas nucleasas dentro de las células también plantea un desafío importante para su eficacia. Además, dirigir la edición a más de un gen (es decir, multiplexación) es difícil con métodos anteriores en comparación con los métodos basados en CRISPR, lo que ahorra tiempo, costo y esfuerzo. La edición genética realizada por CRISPR es heredable, como se demostró en crías de ratas cuando se transfectaron simultáneamente cuatro sgRNAs.
Las tecnologías de secuenciación de ADN y ARN de alto rendimiento han añadido nuevas dimensiones al descubrimiento de mutaciones en la mayoría de las enfermedades humanas. La lista de nuevas enfermedades neurodegenerativas asociadas con cambios genéticos ha crecido rápidamente. Sin embargo, la mayoría de los trastornos neurodegenerativos genéticos no tienen tratamiento curativo hasta la fecha y solo se administra tratamiento sintomático a los pacientes. El complejo mecanismo de muchos de estos trastornos aún no está claro, lo que hace que el desarrollo de nuevas terapias sea un gran desafío. Se requieren métodos altamente efectivos para corregir mutaciones genéticas en enfermedades neurodegenerativas monogénicas. La tecnología de edición genética basada en CRISPR ofrece una doble vía, ya que ayuda a comprender los mecanismos de la enfermedad generando modelos de enfermedad y también funciona como una 'herramienta molecular' para modificar los genes mutados, actuando como una herramienta terapéutica. Esta avanzada tecnología de edición genética se ha utilizado en el desarrollo de modelos animales, modelos celulares, iPSCs (células madre pluripotentes inducidas) y en la modificación de mutaciones causantes en varios trastornos neurodegenerativos.
- ¿Cómo funciona esta tecnología?
- Aplicaciones de la tecnología CRISPR/Cas9
- CRISPR/Cas9 como herramienta terapéutica en neurología
- Ensayos Clínicos
- Desafíos y limitaciones de la tecnología CRISPR-Cas9 en trastornos neurológicos
- Perspectivas Futuras en Trastornos Neurológicos
- Preguntas Frecuentes
- Conclusión
¿Cómo funciona esta tecnología?
El proceso comienza con el diseño de sgRNAs dirigidos contra secuencias de ADN específicas. El sitio diana puede ser una parte codificante o no codificante del genoma. Los sgRNAs deben cumplir dos condiciones: (1) la secuencia diana debe ser única en comparación con el resto del genoma y (2) debe estar adyacente a los sitios PAM. De hecho, los sitios PAM son la señal de unión para la enzima Cas9. Después de diseñar y sintetizar los sgRNAs, estos se transfectan en las células diana junto con la enzima Cas9, ya sea como un complejo de ribonucleoproteína (RNP) o como plásmidos que contienen sus secuencias. Existen varias estrategias para entregar estos plásmidos o complejos RNP en las células huésped, incluyendo vectores virales (virus adenoasociados [AAV] o lentivirus), lipofectaminas (liposomas y nanopartículas) o mediante electroporación (que crea poros temporales en la membrana celular aplicando alto voltaje).
Los plásmidos insertados traducen múltiples copias de sgRNA y Cas9, que interactúan para formar el complejo sgRNA-Cas9 dentro de la célula. La escisión tiene lugar en el dúplex pequeño ARN-ADN, que se forma entre el espaciador del sgRNA y su secuencia de ADN complementaria cerca del sitio PAM. Este ADN escindido se repara utilizando las vías de Reparación Dirigida por Homología (HDR) o Unión de Extremos No Homólogos (NHEJ). El usuario puede decidir qué vía seguir dentro de la célula. Si se requieren inserciones/indels/cambios de marco en el sitio de corte, se sigue el modo de reparación NHEJ. Sin embargo, estos cambios pueden interrumpir el marco de lectura abierto (ORF) del gen, lo que resulta en su activación o represión.
La vía HDR repara la secuencia diana, desde un solo nucleótido hasta un tamaño grande. Utiliza una 'plantilla de reparación' que contiene el cambio deseado en el ADN, la cual debe transfectarse junto con la transfección de sgRNA-Cas9 en la célula. Esta plantilla también lleva una secuencia homóloga aguas arriba y aguas abajo de la secuencia modificada que complementa los extremos cortados y ayuda a las enzimas reparadoras. La ausencia de la secuencia PAM en la plantilla de reparación la protege de la edición por Cas9. La eficiencia de la reparación mediada por HDR es baja y las DSBs son reparadas por el método NHEJ en varios lugares de la escisión. Por lo tanto, las células resultantes son una mezcla de células no editadas (tipo salvaje), editadas por NHEJ y/o editadas por HDR.
Aplicaciones de la tecnología CRISPR/Cas9
La tecnología CRISPR/Cas9 no solo se limita a la corrección de genes, sino que tiene diversas aplicaciones en la investigación biológica y potencialmente terapéutica:
- Inhibición/activación de genes diana: Cas9 tiene dos dominios nucleasa, RuvC y HNH, que cortan las hebras opuestas. La capacidad nucleasa de Cas9 se puede alterar induciendo mutaciones puntuales en estas dos regiones. Esta Cas9 inactiva (dCas9) es capaz de unirse al ADN diana pero no de cortarlo. Si el sitio diana es la región reguladora del gen de interés, el complejo dCas9-ADN bloquea el inicio de la transcripción, lo que lleva a la Inhibición mediada por CRISPR de la transcripción (CRISPRi). Alternativamente, la dCas9 puede fusionarse con potenciadores que conducen a una alta transcripción del gen diana. Esto se conoce como Activación mediada por CRISPR (CRISPRa).
- Modificación epigenética: Los cambios epigenéticos (marcas) no están regulados por códigos genéticos; afectan la expresión génica mediante la modificación reversible de los nucleótidos del ADN y las proteínas histonas. Las marcas epigenéticas incluyen la adición o eliminación de grupos metilo, acetilo, ubiquitina y fosfato a nucleótidos/histonas. La dCas9 se ha fusionado con éxito con varios modificadores epigenéticos como p300 (histona acetiltransferasa) y LSD1 (lisina-específica desmetilasa 1) para localizar diversas marcas epigenéticas. Estas herramientas no crean DSBs, sino que modifican los nucleótidos del ADN (por ejemplo, citosina a metil citosina) o proteínas histonas (histona H3), activando o desactivando el gen diana.
- Multiplexación: Esto se logra transfectando más de un sgRNA a las células diana. Estos sgRNAs pueden clonarse en un único plásmido o el número requerido de sgRNAs (como RNPs) puede entregarse directamente en las células. El plásmido multiplicará los sgRNAs dentro de las células, mientras que los RNPs actuarán directamente en los sitios diana. Esto, a su vez, aumenta la edición del objetivo. Los sitios diana de estos múltiples sgRNAs pueden ser únicos o múltiples, e incluso se puede apuntar a la deleción/edición de grandes regiones del genoma eliminando la secuencia entre dos sitios diana. Se ha reportado la multiplexación de entre 2 y 7 loci génicos clonando sgRNAs en un solo plásmido.
- Barridos a nivel genómico usando CRISPR: Esto se realiza generando una gran población de células que contienen la mutación deseada o el estado activado/reprimido de uno o varios genes de interés. Posteriormente, estas células se utilizan para identificar las perturbaciones genéticas que resultan en fenotipos deseados. Los barridos genéticos directos son particularmente útiles para estudiar fenotipos diversos cuya causa subyacente no se conoce. La tecnología CRISPR es capaz de realizar modificaciones genéticas altamente específicas y permanentes que anulan las funciones de los genes diana. Esta tecnología ya se ha utilizado ampliamente en el cribado de nuevos genes que regulan fenotipos conocidos involucrados en la resistencia a fármacos, la viabilidad celular y los sitios de tumores metastásicos.
- Visualización de loci genómicos usando fluoróforos: Aquí, la dCas9 se etiqueta con un marcador fluorescente como GFP (proteína fluorescente verde). Estos sgRNAs etiquetados se unen al sitio genómico diana y emitirán fluorescencia cuando se irradien con luz de una longitud de onda específica. Las células vivas también pueden visualizarse usando imágenes CRISPR. Múltiples loci genéticos pueden ser dirigidos con la tecnología fácil de usar de diseño de sgRNA. La hibridación fluorescente in situ (FISH) también puede integrarse con CRISPR para revelar la dinámica de la cromatina en células vivas. Se puede usar más de un tipo de fluoróforo, haciéndolo multicolor. Si se dirige todo el cromosoma mediante múltiples sgRNAs etiquetados con fluoróforos, se denomina 'pintura cromosómica'.
- Purificación de regiones genómicas: La expresión de cualquier gen está regulada por varios factores de transcripción (TFs) que se unen a las regiones reguladoras. Identificar estas secuencias (ADN) y TFs y modular sus características puede ayudar a controlar la expresión de esos genes. Al igual que en las imágenes CRISPR, etiquetar la dCas9 con biotina, epítopos (3×FLAG) o anticuerpos anti-Cas9 puede usarse para unirse a cualquier secuencia génica diana (génica o intergénica). La dCas9 unida a etiquetas puede ser arrastrada por sus etiquetas adjuntas. Después de la purificación, la fracción unida puede identificarse mediante secuenciación de próxima generación (ADN) o espectrometría de masas (TFs). Esta purificación basada en CRISPR es muy útil para identificar ADN de interés y sus proteínas unidas (TFs) in vitro.
CRISPR/Cas9 como herramienta terapéutica en neurología
Numerosas enfermedades neurodegenerativas han sido objeto de la tecnología CRISPR, ya sea para desarrollar modelos de enfermedad o para fines terapéuticos. El primer informe sobre el uso de esta tecnología en enfermedades neurodegenerativas fue en la enfermedad priónica, donde se eliminó el gen PRP para anular su expresión en líneas celulares. A continuación, destacamos el estado actual de la tecnología CRISPR-Cas9 en los trastornos neurológicos más comunes:
Enfermedad de Alzheimer
La Enfermedad de Alzheimer (EA) es un trastorno neurodegenerativo progresivo y el más común, conocido como la principal causa de muerte entre las personas mayores. Aunque la mayoría de los casos de EA son esporádicos, varias mutaciones genéticas se han relacionado con su aparición. Mutaciones en presenilinas (PSEN1 y PSEN2) y en la proteína precursora de amiloide (APP) se relacionan con el inicio temprano, mientras que mutaciones en apolipoproteína-E (ApoE) se asocian con el inicio tardío. La escisión anormal de la proteína APP por la enzima β-secretasa-1 (codificada por el gen BACE1) es crucial para la producción del péptido amiloide β (Aβ) y las placas amiloides, características de la EA.
Se observó una reducción significativa en la acumulación de placas amiloides y una disminución en la secreción de Aβ de neuronas postmitóticas del hipocampo cuando el gen BACE1 fue dirigido a través de nanopartículas recubiertas con el complejo Cas9-sgRNA en modelos de ratón KI (knock-in) de EA. Múltiples entregas de nanopartículas en la región del hipocampo demostraron ser más efectivas para suprimir el gen BACE1. Anteriormente, en 2018, se inactivaron (KO) dos mutaciones suecas en fibroblastos humanos usando tecnología CRISPR, reduciendo los niveles de la proteína APP mutante sin cambiar su forma de tipo salvaje. Posteriormente, esto se intentó en modelos de ratón portadores de la misma mutación, donde se usaron virus (vectores) para entregar el complejo CRISPR-Cas9 directamente en el hipocampo de ratones adultos, obteniendo resultados similares a los del estudio in vitro. Este fue el primer estudio de entrega viral de CRISPR in vivo en modelos de EA. Recientemente se ha logrado el rescate exitoso de mutaciones en PSEN1 mediante CRISPR, así como la inactivación de APP en neuronas corticales derivadas de células madre humanas.
El polimorfismo de KIBRA (proteína expresada en riñón y cerebro) está bien asociado con el rendimiento cognitivo en la EA. Un grupo de investigación desarrolló recientemente un ratón KIBRA-KO basado en CRISPR para investigar su papel en la EA. Encontraron una alta pérdida neuronal en ratones KIBRA-KO en el hipocampo por apoptosis, mientras que la sobreexpresión de KIBRA en líneas celulares neuronales promovió significativamente su proliferación e inhibió la apoptosis inducida por Aβ. Esto demostró que KIBRA funciona como un gen neuroprotector que promueve la supervivencia celular e inhibe la apoptosis inducida por Aβ, convirtiéndolo en un objetivo terapéutico potencial para el tratamiento de la EA.
Enfermedad de Parkinson
La Enfermedad de Parkinson (EP) es un trastorno neurodegenerativo progresivo causado por la muerte de las neuronas dopaminérgicas en la sustancia negra. Se han establecido causas tanto esporádicas como genéticas, como mutaciones en los genes PINK1, SNCA (α-sinucleína) y LRRK2, en la EP. La característica distintiva de la EP es la presencia de cuerpos de Lewy llenos de proteína α-sinucleína mutante en neuronas moribundas o muertas. Múltiples copias del gen SNCA aceleran la sinucleopatía, causando una enfermedad grave. Se utilizó CRISPRi para reducir la expresión del gen SNCA en líneas celulares neuronales y otras, encontrando una alta supresión de hasta el 60%. Posteriormente, otro estudio intentó modular las marcas de metilación (epigenéticas) ubicadas en el intrón 1 del gen SNCA que controla su expresión. Utilizaron un vector viral 'todo en uno', es decir, dCas9 fusionada con el dominio catalítico de la enzima ADN metiltransferasa-3A, para transfectar neuronas dopaminérgicas derivadas de iPSCs que contenían copias triplicadas del gen SNCA. Se observaron niveles reducidos de ARNm y proteína SNCA y se mejoraron otros parámetros como el ROS mitocondrial y la viabilidad celular.
Los canales de calcio activados en neuronas (MCU) y la disfunción mitocondrial mediada por calcio también estimulan la muerte de las neuronas dopaminérgicas en la EP. En un estudio, se utilizó un modelo de pez cebra de EP con el gen PINK eliminado (PINK–/–) que mostraba un funcionamiento mitocondrial anormal. Cuando se desactivó el gen MCU induciendo una mutación de cambio de marco en su exón 3 de este modelo de EP utilizando tecnología CRISPR, se restauró el funcionamiento mitocondrial. Esto demostró efectos protectores de la inactivación del gen MCU en modelos de EP y proyecta este gen para el desarrollo de nuevas terapias.
Recientemente, se ha desarrollado otra línea de iPSCs de EP que contiene el gen tirosina hidroxilasa (TH) etiquetado con GFP utilizando la técnica CRISPR/Cas9. La enzima TH realiza el paso limitante de la velocidad durante la biosíntesis de dopamina y se ha establecido como un marcador confiable para las neuronas dopaminérgicas. Esta construcción génica etiquetada con GFP fue muy útil para monitorear la eficiencia de la transfección. En otro informe, se desarrollaron astrocitos derivados de iPSCs que contenían una mutación en el gen LRRK2, reportada en casos de EP autosómica dominante. Estos astrocitos de EP mostraron maquinaria autofágica deteriorada y acumulación de α-sinucleína endógena.
Organismos superiores como cerdos y chimpancés comparten una fisiología celular muy similar con los humanos. Los beneficios de usar modelos animales de enfermedades como estos son que procesan las proteínas enfermas de manera casi idéntica y producen síntomas y pronósticos aproximadamente iguales a los de los humanos. Se ha generado un modelo de EP en minicerdos Guangxi Bama, en el que se introdujeron tres mutaciones en SCNA utilizando tecnología CRISPR. Aunque los animales genéticamente modificados no mostraron síntomas de enfermedad a los 3 meses de edad, el envejecimiento podría introducir síntomas de enfermedad en estos animales.
Enfermedad de Huntington
La Enfermedad de Huntington (EH) es otro trastorno neurodegenerativo genético bien conocido causado por la sobreexpansión de repeticiones de citosina-adenina-guanina (CAG) en el gen huntingtina (HTT). Es un fenotipo autosómico dominante, lo que significa que la expansión heterocigota causará el fenotipo, convirtiendo estas repeticiones en objetivos terapéuticos utilizando la tecnología CRISPR-Cas9.
Se llevó a cabo la deleción basada en CRISPR de una región de 44 kb que incluía el promotor, el sitio de inicio de la transcripción y la mutación de expansión de CAG del gen HTT mutante para inactivar completamente el alelo mutante. Esto resultó en la inhibición completa del ARNm y la proteína mHTT sin afectar el alelo normal. En otro estudio, se dirigieron la región 5′-UTR (región no traducida) y el límite exón-1-intrón mediante el método CRISPR-Cas9 para interferir con su transcripción. La entrega basada en plásmidos de CRISPR-Cas9 se realizó en células madre mesenquimales (MSCs) extraídas de la médula ósea de ratones YAC128, un modelo de EH. La traducción de la proteína HTT mutante se redujo en las células MSC, lo que representa un paso hacia la terapia de la EH. Un protocolo más reciente basado en el uso de Cas9 nickasa (que corta solo una hebra del ADN diana y no produce DSB) para cortar y reemplazar las repeticiones de CAG expandidas con repeticiones de CAG normales ha mostrado una reducción de hasta el 70% en los niveles de proteína. En otro informe, la entrega de Cas9 mediante AAV se realizó directamente en el cuerpo estriado para interrumpir la HTT mutante en el modelo de ratón de EH, R6/2. La interrupción de la proteína mutante redujo la formación de inclusiones neurotóxicas en dos veces y mejoró los déficits motores, lo que llevó a una vida más larga del animal en comparación con los ratones no transfectados.
Ataxia de Friedreich
La Ataxia de Friedreich (AF) es un fenotipo autosómico recesivo causado por la expansión homocigota de trinucleótidos guanina-adenina-adenina (GAA) en el primer intrón del gen FXN, que codifica la frataxina, una proteína mitocondrial crucial. Los niveles de proteína frataxina están inversamente relacionados con la gravedad de la enfermedad. Es un trastorno de inicio temprano caracterizado por ataxia de la marcha, dificultad para hablar y tragar, y cardiomiopatía hipertrófica, que lleva al paciente a la silla de ruedas y a la muerte a principios de los 20 años.
Se realizó la entrega sistémica de vectores virales en modelos de ratón YG8sR y YG8R de AF. Estos modelos de ratón contienen una y dos copias en tándem del transgén FXN humano, respectivamente. En ratones YG8sR transfectados con CRISPR-Cas9, los niveles de proteína frataxina aumentaron, mientras que en ratones YG8R, los niveles de proteína disminuyeron. Esto podría explicarse por la presencia de dos copias del gen FXN en los últimos; la deleción de una copia llevó a una expresión proteica reducida. Sin embargo, la entrega exitosa del complejo Cas9 en modelos de ratón abrió un camino prometedor para futuras terapias de la AF.
Recientemente, se ha desarrollado un nuevo modelo celular inducible de AF, HEK-CFXN, mediante la inactivación del gen FXN endógeno de células HEK293 a través del método CRISPR-Cas9 y la adición de un gen FXN 'inducible'. Se pudieron producir diferentes niveles de proteína frataxina activándolo. Curiosamente, este estudio también demostró que la sobreexpresión de proteína frataxina podría conducir a alto estrés oxidativo y pool de hierro lábil, que fueron tan tóxicos como en células de pacientes con AF. Sin embargo, este nuevo modelo celular proporciona un modelo celular útil para investigar más a fondo el papel de la frataxina en la fisiología celular.
Síndrome de X Frágil
El Síndrome de X Frágil (SXF) es un fenotipo dominante ligado al cromosoma X causado por la expansión de repeticiones de trinucleótidos citosina-guanina-guanina (CGG) en la región 5′-UTR del gen FMR1. Los alelos de mutación completa contienen más de 200 repeticiones de CGG y la hipermetilación aguas arriba de estas repeticiones conduce a una pérdida de la proteína codificada por FMR1 (FMRP). La presencia de una expansión de repetición anormal aguas arriba de la región codificante proporciona una buena oportunidad para editar estas secuencias y desarrollar terapias. El estado hipermetilado de la secuencia aguas arriba se corrigió espontáneamente (es decir, desmetiló) cuando se escindieron las repeticiones de CGG tanto en células madre embrionarias como en iPSCs derivadas de pacientes con SXF. La expresión de FMRP se restauró y mantuvo en células neuronales precursoras y maduras. La restauración de FMRP también se logró en una línea celular híbrida somática y en iPSCs derivadas de pacientes cuando se escindieron estas repeticiones de CGG patogénicas utilizando tecnología CRISPR. En otro estudio, se etiquetó la Cas9 con un activador transcripcional VP192 con el objetivo de mejorar la transcripción de FMR1, que se reduce patogénicamente debido a la expansión de las repeticiones de CGG. Este complejo CRISPR-Cas9 etiquetado fue capaz de aumentar los niveles de FMRP en un modelo de SXF de hESCs (células madre embrionarias humanas).
La entrega basada en nanopartículas del complejo CRIPSR-Cas9 (RNP), llamado CRISPR-gold, se utilizó para transfectar neuronas hipocampales cultivadas primariamente de modelos de ratón de SXF. La citotoxicidad fue muy baja y la transfección no mostró efectos adversos en la salud de la membrana neuronal o la excitabilidad neuronal. Posteriormente, el mismo CRISPR-gold se utilizó para entregar el complejo RNP dirigido al gen Grm5 dentro del cerebro de ratones FMR1–/– (deficientes en el gen FMR1), un modelo de ratón de SXF. El gen Grm5 codifica el receptor metabotrópico de glutamato 5 (mGluR5), que está exagerado y se ha asociado con la fisiopatología del SXF. Los ratones transfectados mostraron una reducción en los niveles de mGluR5 (40%-50%) sin efectos adversos significativos. Los cambios de comportamiento (comportamiento excesivo de excavación y salto) también se normalizaron en estos ratones. Recientemente, se ha generado una línea de hESCs FMR1-KO homocigota utilizando la tecnología CRISPR-Cas9 que contiene una deleción homocigota de 280 nucleótidos en el exón 1, eliminando el codón de inicio. Esta línea de hESC fue capaz de diferenciarse y retuvo la pluripotencia y el cariotipo normal.
Ataxias Espinocerebelosas
La Ataxia Espinocerebelosa tipo 2 (AEC2) es un trastorno autosómico dominante causado por la expansión de trinucleótidos (CAG) en el primer exón del gen ATXN2. Los pacientes con AEC2 desarrollan ataxia de la marcha, visión doble y problemas del habla con el tiempo. Un grupo de investigación utilizó la tecnología CRISPR-Cas9 para generar líneas celulares 'corregidas' de AEC2. Reemplazaron las repeticiones patogénicas (CAG) (n = 36) para desarrollar una línea de iPSCs de AEC2 y la llamaron H196. Estas células corregidas mostraron cariotipo normal y no portaban ningún cambio de marco u otra mutación. Utilizaron células de otros pacientes y de manera similar desarrollaron dos líneas celulares iPSC más, H271 y H266, en las que reemplazaron las repeticiones (CAG)44 con repeticiones normales (CAG)22. Estas líneas celulares proporcionarán nuevas y verdaderas líneas celulares iPSC de control para la investigación de la AEC2.
En la Ataxia Espinocerebelosa tipo 3 (AEC3), se reportó la deleción de repeticiones patogénicas (CAG)78 en el gen ATX3 a partir de iPSCs derivadas de pacientes. Las células corregidas retuvieron la pluripotencia y pudieron diferenciarse aún más en células neuronales.
Distrofia Muscular de Duchenne
La Distrofia Muscular de Duchenne (DMD) es una enfermedad neuromuscular grave y muy prevalente. Diagnosticada entre los 3 y 5 años de edad, comienza con el debilitamiento de los músculos del tronco y las extremidades inferiores y progresa con marcha anormal y retraso motor. A los 10 años de inicio, los pacientes quedan en silla de ruedas. Es una enfermedad recesiva ligada al cromosoma X y, por lo tanto, se limita a los niños. El gen DMD, distrofina (DMD), es el gen más largo conocido, conteniendo 2.3 × 106 pares de bases. Se han reportado más de 4000 mutaciones hasta la fecha, de las cuales dos tercios son grandes deleciones que se extienden sobre los exones 2–20 y 44–53. El gen DMD ha sido dirigido in vitro e in vivo por diversas tecnologías en numerosos estudios. La mayoría de estos informes se centraron en restaurar las regiones mutadas en los exones.
La función de la proteína DMD se restauró hasta un 18% en el modelo de ratón mdx de DMD (los ratones mdx poseen una mutación en el exón 23). Se probaron tanto la variación NHEJ como HDR de CRISPR en estos estudios con el objetivo de normalizar la mutación cC3185T del exón 23 en el modelo de ratón mdx. Además, se obtuvo una restauración de la proteína diana de hasta el 80% de los ratones de tipo salvaje en otro modelo de ratón de DMD que carecía del exón 50. Se utilizó la entrega viral (AAV) del complejo Cas9-gRNA para lograr esta alta frecuencia de restauración. Se inyectaron nanopartículas recubiertas de oro con CRISPR (CRISPR-gold) intramuscularmente en ratones mdx. Los resultados mostraron que los niveles de distrofina se restauraron y la fuerza muscular también mejoró. Un estudio reciente utilizó una nueva nucleasa Cas9, Cpf1, para corregir el gen DMD mutado en iPSCs de un modelo de ratón. La nucleasa Cpf1 se diferencia de Cas9 en que produce extremos romos y requiere PAMs ricos en T.
Distrofia Miotónica
La Distrofia Miotónica (DM) es la distrofia más común entre los adultos, causada por una mutación de expansión de nucleótidos en los genes causantes. La expansión anormal de repeticiones de trinucleótidos citosina-timina-guanina (CTG) en la región 3′-UTR del gen DMPK en el cromosoma 19 causa la distrofia miotónica tipo 1 (DM1), mientras que la expansión de tetranucleótidos (CCTG) en el intrón 1 del gen CNBP conduce a la distrofia miotónica tipo 2 (DM2). La enfermedad comienza con el debilitamiento y desgaste de los músculos, miotonía y dificultades en la respiración. Las expansiones de nucleótidos patogénicas han sido objetivos de las herramientas de edición genética en los genes DMPK/CNBP. Curiosamente, la escisión completa del gen DMPK no afectó la supervivencia de las células. Las células editadas (mioblastos) no mostraron signos de focos ribonucleares patogénicos en sus núcleos. Más tarde, la dCas9 modificada logró eliminar el ARNm mutado, lo que resultó en una reducción de hasta el 90% en mioblastos de DM1. En otro estudio, la dCas9 se entregó mediante vectores virales (AAV) en la vena temporal de un modelo de ratón de DM1. Esto llevó a una mejora de la miotonía en la mayoría de los ratones.
Ensayos Clínicos
Los ensayos clínicos basados en CRISPR aún no han comenzado específicamente para enfermedades neurodegenerativas. La mayoría de los ensayos clínicos en curso se centran en la inmunoterapia para el cáncer. La edición basada en CRISPR de los receptores de antígenos de células T (tecnología CAR-T) se está probando para el tratamiento del melanoma, el sarcoma y el mieloma múltiple en EE. UU. Otro ensayo clínico de CAR-T ha comenzado para la leucemia y el linfoma de células B, el carcinoma de pulmón de células no pequeñas (CPCNP) metastásico y el cáncer de vejiga invasivo en China. Además de los cánceres, un tratamiento ocular basado en CRISPR-Cas9 inyectable localmente para la amaurosis congénita de Leber (ACL) está en fase de ensayo clínico. La mutación más común en el gen causante CEP20 es el objetivo de Cas9 con el fin de mejorar la función de las células fotorreceptoras antes de que la enfermedad progrese a la pérdida de visión.
Desafíos y limitaciones de la tecnología CRISPR-Cas9 en trastornos neurológicos
Aunque es una tecnología de edición genética comparativamente nueva y más avanzada, presenta varios contratiempos. El primero es el propio genoma huésped. Los sitios a modificar pueden tener suficiente similitud de secuencia con otras partes del genoma, lo que lleva a cortes no deseados en el genoma huésped. Así, se generan mutaciones no deseadas, que pueden afectar adversamente la salud general e incluso la supervivencia del organismo huésped. La entrega in vivo es otro factor limitante. Cruzar la barrera hematoencefálica (BHE) es un desafío primordial para los componentes de esta tecnología de edición genética. Aunque ya se han desarrollado varios mecanismos (mencionados anteriormente), la seguridad del huésped es una preocupación importante. La entrega mediada por virus in vivo sigue siendo cuestionable, ya que los efectos a largo plazo del uso de dichos virus animales y las ramificaciones de desmontar los componentes CRISPR en su efectividad en el cerebro aún están bajo investigación. La entrega precisa y segura a los tejidos diana, respetando los tejidos sanos, también es una preocupación importante para la transfección basada en liposomas/nanopartículas de los componentes CRISPR-Cas9. Además, la edición genética mediada por HDR requiere una maquinaria de reparación de ADN eficiente, que puede ser menos activa en células postmitóticas como las neuronas. La corrección genética específica de alelos es otra gran preocupación a superar. Aunque se han realizado varios intentos para corregir el alelo patogénico en la EH, otros trastornos como los tipos de AEC -1, -2 y -3 plantean un gran desafío para la implementación de esta tecnología en entornos clínicos.
La respuesta inmune a CRISPR-Cas9 y la presencia de anticuerpos preexistentes contra Cas9 (una proteína) podrían ser un obstáculo significativo, especialmente para la edición genética in vivo. Es muy deseable realizar una evaluación más rigurosa de las posibles respuestas inmunológicas al origen microbiano del sistema. Sin embargo, la entrega de complejos RNP y la optimización de codones pueden ser clave para superar tales obstáculos.
| Tecnología | Precisión | Eficiencia | Costo | Multiplexación | Efectos Fuera del Objetivo |
|---|---|---|---|---|---|
| CRISPR-Cas9 | Alta | Alta | Bajo/Moderado | Fácil | Presentes (mejorando) |
| TALENs | Alta | Moderada | Alto | Difícil | Presentes |
| ZFNs | Moderada/Alta | Baja/Moderada | Alto | Difícil | Presentes |
Perspectivas Futuras en Trastornos Neurológicos
Se han encontrado varios polimorfismos de un solo nucleótido (SNPs) en diferentes enfermedades neurodegenerativas. El papel de dichos SNPs puede investigarse combinando dCas9 con enzimas modificadoras de nucleótidos, como la citidina deaminasa o la adenosina deaminasa, que convierten el nucleótido simple en su derivado sin causar DSBs. La remodelación de promotores, potenciadores y represores que deciden los niveles de los genes necesita más investigación utilizando diferentes aplicaciones de la tecnología CRISPR, como CRISPRi, CRISPRa o dCas9. El papel de las moléculas no codificantes, como los microARNs, los ARNs que interactúan con Piwi (piRNAs) y los ARNs largos no codificantes (ncRNAs), debe investigarse más a fondo, ya que se han involucrado en varias enfermedades neurodegenerativas. Además, se deben derivar nuevos modelos animales que expresen las enfermedades con mayor precisión a partir de iPSCs generadas con CRISPR-Cas9 de enfermedades neurodegenerativas, lo que se ha llevado a cabo para crear organoides renales e intestinales derivados de iPSCs.
Por encima de todo, los efectos fuera del objetivo que induce Cas9 al unirse y cortar secuencias no diana deben abordarse mejorando la eficiencia de Cas9. Sin embargo, también se han investigado nuevas enzimas de edición genética como Cpf1, Cas12a y CasX, que poseen una mejor eficiencia de edición y menores efectos fuera del objetivo en diversas enfermedades.
Preguntas Frecuentes
- ¿Qué es CRISPR-Cas9 en neurología?
- CRISPR-Cas9 es una tecnología de edición genética que permite modificar secuencias de ADN específicas. En neurología, se utiliza para estudiar las causas genéticas de enfermedades neurodegenerativas, crear modelos celulares y animales de estas enfermedades y, potencialmente, corregir las mutaciones genéticas subyacentes.
- ¿Se utiliza CRISPR en neurociencia?
- Sí, la tecnología CRISPR-Cas9 se utiliza ampliamente en neurociencia para entender la función de los genes en el cerebro, modelar enfermedades neurológicas, desarrollar terapias génicas, realizar cribados genéticos y manipular la actividad neuronal.
- ¿Qué enfermedades cura el CRISPR?
- Actualmente, CRISPR-Cas9 no ha curado ninguna enfermedad neurológica en humanos. Está en etapas de investigación preclínica y en algunos ensayos clínicos para otras enfermedades (principalmente cáncer y una enfermedad ocular). Su potencial para curar enfermedades neurológicas es muy prometedor, pero aún enfrenta desafíos significativos antes de ser una terapia establecida.
- ¿Hay ensayos clínicos con CRISPR para enfermedades neurológicas?
- Hasta la fecha, no hay ensayos clínicos en curso específicamente para tratar enfermedades neurodegenerativas utilizando CRISPR-Cas9. Los ensayos clínicos actuales se centran principalmente en el cáncer y una enfermedad ocular, la amaurosis congénita de Leber.
Conclusión
Las ventajas de la tecnología CRISPR-Cas9, como su alta precisión, fácil personalización, mejor multiplexación y rentabilidad en comparación con otros métodos convencionales de edición genética, se han demostrado en numerosos estudios. Sin embargo, la entrega de las partes de sgRNAs y Cas9 dentro de las células diana y los efectos fuera del objetivo asociados siguen siendo desafíos para el éxito de este método. Aunque esta tecnología ha revelado varios mecanismos patogénicos nuevos en trastornos neurológicos y se han preparado varios modelos de enfermedad, la modificación del gen defectuoso en trastornos monogénicos aún no se ha logrado completamente en muchos casos. Dirigir la secuencia huésped en un tipo celular particular en tejido/órgano y reducir los efectos fuera del objetivo es difícil, pero será alcanzable en un futuro cercano utilizando enzimas de edición de versión más avanzada. Sin embargo, la edición de la secuencia de un gen es irreversible y, por lo tanto, se deben tomar precauciones extremas para minimizar los cambios genéticos dañinos, programando el momento de la entrega de CRISPR-Cas9 y su dosificación. Estos avances allanarán el camino para que la tecnología CRISPR-Cas9 se establezca en la clínica rutinaria, brindando una oportunidad para salvar vidas.
Si quieres conocer otros artículos parecidos a CRISPR-Cas9: Revolución Genética en Neurología puedes visitar la categoría Neurociencia.