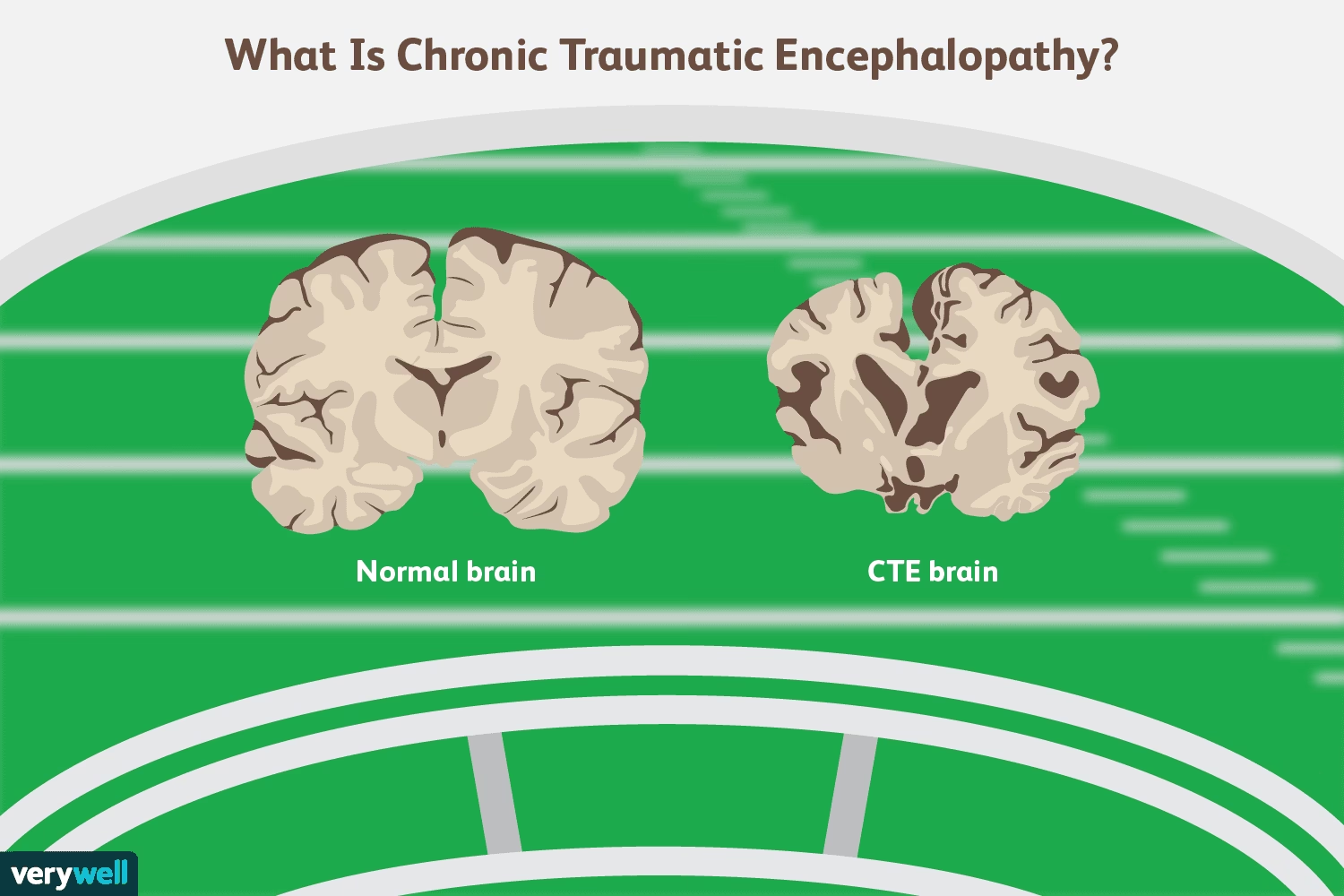
La encefalopatía traumática crónica (ETC) es una condición neurodegenerativa progresiva asociada con el trauma craneal repetitivo. Si bien se reconoce con mayor frecuencia en atletas que participan en deportes de contacto, también se ha informado en individuos expuestos a lesiones por explosión y otras formas de neurotrauma recurrente. Los síntomas clínicos de la ETC son variados e incluyen cambios en el comportamiento y el estado de ánimo, pérdida de memoria, deterioro cognitivo y, en casos graves, demencia. A diferencia de muchas otras enfermedades neurológicas que pueden sospecharse clínicamente, el diagnóstico definitivo de la ETC solo es posible mediante el examen neuropatológico post mortem del tejido cerebral.
Históricamente, la conexión entre el trauma craneal repetido y una condición neurológica crónica se reconoció por primera vez hace casi un siglo en boxeadores, inicialmente descrita como "punch drunk" por Harrison Martland en 1928. Con el tiempo, se introdujeron otros términos como "encefalopatía traumática progresiva" y "demencia pugilística". La designación más amplia de "encefalopatía traumática crónica" (ETC) se popularizó a partir de la década de 1940, reconociendo que la condición podía surgir de diversas fuentes de trauma, no solo el boxeo.
Los primeros informes neuropatológicos detallados se centraron en boxeadores retirados que desarrollaban parkinsonismo y demencia. Los hallazgos macroscópicos incluían atrofia cerebral, ventrículos agrandados, cavum septum pellucidum y despigmentación de la sustancia negra. A nivel microscópico, se observaban ovillos neurofibrilares (ONF) en la corteza y el tronco encefálico. Un estudio seminal de Corsellis y colaboradores en 1973 en 15 boxeadores amplió estos hallazgos, describiendo ventrículos laterales y tercer ventrículo agrandados, adelgazamiento del cuerpo calloso, cavum septum pellucidum fenestrado y cicatrización de las amígdalas cerebelosas. Microscópicamente, notaron pérdida neuronal y degeneración neurofibrilar. Estudios posteriores reexaminando estas muestras confirmaron la presencia de depósitos de beta-amiloide (Aβ) en un alto porcentaje de casos de boxeadores, aunque la relación de Aβ con la ETC ha sido objeto de debate y parece estar más asociada a la edad en poblaciones más amplias de ETC.
La comprensión de la ETC se expandió para incluir a mujeres y personas sin antecedentes de boxeo. En 1990, se reportó el primer caso en una mujer con antecedentes de abuso físico, y en 1991, en una mujer joven con comportamientos de auto-agresión (golpearse la cabeza). Estos casos mostraron hallazgos neuropatológicos similares, aunque con variaciones en la distribución y la ausencia de Aβ en el caso más joven.
Un avance crucial en la definición de la ETC se produjo con los estudios que identificaron la patología característica en individuos más jóvenes y en participantes de deportes modernos como el fútbol americano, el fútbol soccer y el hockey sobre hielo. Geddes y colaboradores en 2004 reportaron hallazgos en cerebros de cinco hombres jóvenes con antecedentes de trauma craneal repetitivo, incluyendo un jugador de fútbol soccer y un boxeador. Aunque no había cambios macroscópicos evidentes, el examen microscópico reveló ONF y neuritas neuropilares positivos para tau hiperfosforilada (p-tau), dispuestas en clusters llamativos alrededor de pequeños vasos sanguíneos intracorticales, a menudo en las profundidades de los surcos corticales. Esta distribución era marcadamente diferente de la patología inicial de la enfermedad de Alzheimer (EA). Omalu y colaboradores describieron hallazgos similares en jugadores de la NFL y un luchador profesional, consolidando la asociación de esta patología distintiva con el trauma craneal repetitivo en diversas poblaciones.
- Hallazgos Neuropatológicos Clave en la ETC
- Hallazgos Macroscópicos (Anatomía Patológica Bruta)
- Hallazgos Microscópicos Detallados y Criterios Diagnósticos
- Estadificación de la Patología de Tau en la ETC
- Patología de TDP-43 en la ETC
- Beta-Amiloide (Aβ) en la ETC
- Comorbilidades Neurodegenerativas
- Relación entre Trauma Craneal Repetitivo y Patología
- Propagación de la Patología de Tau
- Patología Axonal
- Preguntas Frecuentes sobre la Neuropatología de la ETC
Hallazgos Neuropatológicos Clave en la ETC
El sello distintivo de la ETC a nivel microscópico es la deposición anormal de la proteína tau hiperfosforilada (p-tau). Esta proteína se acumula en forma de ovillos neurofibrilares (ONF) dentro de las neuronas, así como en astrocitos (ovillos astrocíticos) y en las prolongaciones neuronales (neuritas distróficas). Lo más característico es que estas acumulaciones de p-tau se presentan en grupos distintivos alrededor de pequeños vasos sanguíneos en la corteza cerebral, particularmente concentrados en las profundidades de los surcos corticales.
En los casos leves, la patología de p-tau puede ser focal, limitada a estos epicentros perivasculares en las profundidades de los surcos. A medida que la enfermedad progresa, la patología de p-tau se extiende, afectando capas corticales más superficiales (especialmente las capas II y III) y propagándose a otras regiones del cerebro, incluyendo el lóbulo temporal medial (hipocampo, amígdala, corteza entorrinal), diencéfalo, tronco encefálico y cerebelo.
Además de la patología de tau, la acumulación anormal de la proteína TDP-43 de 43 kDa se encuentra en la mayoría de los casos de ETC. Esta patología puede variar desde neuritas citoplasmáticas hasta inclusiones neuronales y gliales. La patología de TDP-43 tiende a ser más prominente en las etapas avanzadas de la ETC y puede solaparse en distribución con la que se observa en la degeneración lobar frontotemporal con TDP-43 (DLFT-TDP), sugiriendo posibles mecanismos patogénicos compartidos. La presencia de TDP-43 en la ETC es importante porque muchas enfermedades causadas por alteraciones en el metabolismo de TDP-43 también muestran alteraciones en el metabolismo de tau.
La deposición de beta-amiloide (Aβ), la proteína clave en la enfermedad de Alzheimer, se identifica en aproximadamente el 43% de los casos de ETC en series amplias, y está significativamente asociada con la edad al momento del fallecimiento. Aunque la Aβ puede estar presente y contribuir a la carga patológica y clínica, no es un hallazgo consistente ni definitorio de la ETC, a diferencia de la patología de p-tau perivascular.
Hallazgos Macroscópicos (Anatomía Patológica Bruta)
En las etapas tempranas o leves de la ETC, los cambios macroscópicos en el cerebro suelen ser sutiles o inexistentes. Los hallazgos más comunes en estas fases pueden incluir un cavum septum pellucidum (una cavidad entre las láminas del septum pellucidum) y un ligero agrandamiento de los cuernos frontales y temporales de los ventrículos laterales. También pueden observarse espacios perivasculares prominentes en la sustancia blanca.
En las etapas intermedias y avanzadas de la ETC, los cambios macroscópicos se vuelven más pronunciados y característicos. Estos incluyen:
- Reducción del peso cerebral.
- Atrofia de la sustancia gris y blanca, típicamente más grave en los lóbulos frontales y temporales anteriores.
- Agrandamiento de los ventrículos laterales y el tercer ventrículo.
- Cavum septum pellucidum, a menudo con fenestraciones (perforaciones) en el septum.
- Atrofia del tálamo, hipotálamo y cuerpos mamilares.
- Adelgazamiento del istmo del cuerpo calloso.
- Despigmentación del locus coeruleus y la sustancia negra.
A diferencia de los informes iniciales centrados en boxeadores, las anomalías macroscópicas cerebelosas son menos comunes en la ETC asociada con otros deportes o actividades.
Hallazgos Microscópicos Detallados y Criterios Diagnósticos
La característica microscópica definitoria de la ETC es la presencia de depósitos de p-tau en clusters distintivos. Estos clusters se localizan predominantemente alrededor de pequeños vasos sanguíneos (distribución perivascular) y en las profundidades de los surcos corticales. La patología de p-tau incluye ONF, pre-ovillos y neuritas distróficas. En los casos leves, esta patología es focal, mientras que en etapas más avanzadas se extiende a capas corticales más superficiales (capas II y III) y otras regiones cerebrales.
Los ovillos astrocíticos subpiales en las profundidades de los surcos son otro hallazgo característico, incluso en ausencia de patología neuronal perivascular subyacente, y no se encuentran en cerebros envejecidos sin antecedentes de trauma craneal repetitivo.
Se han propuesto criterios provisionales para el diagnóstico neuropatológico de la ETC, que actualmente están siendo validados. Estos criterios se basan en la hipótesis de que la patología de p-tau irregular y perivascular en clusters en las profundidades de los surcos corticales distingue la ETC de otras taupatías como la enfermedad de Alzheimer. Los criterios incluyen (adaptado de McKee et al.):
- Focos perivasculares de ONF, pre-ovillos y neuritas dot-like y thread-like con inmunorreactividad a p-tau en la neocorteza.
- Distribución irregular de ONF, ovillos astrocíticos (OAs) y neuritas con inmunorreactividad a p-tau en las profundidades de los surcos cerebrales, a menudo junto a vasos sanguíneos penetrantes.
- ONF en las crestas de la corteza cerebral localizados preferentemente en las capas superficiales II y III, una característica especialmente prominente en la neocorteza temporal.
- Clusters de OAs subpiales en la corteza cerebral, más pronunciados en las profundidades de los surcos.
- OAs subependimarios en las regiones periventriculares de los ventrículos laterales, sustancia gris periacueductal y tronco encefálico lateral.
Las isoformas de tau (3R y 4R) se encuentran en la patología neuronal de la ETC, similar a la EA. Sin embargo, la isoforma 4R de tau predomina en los astrocitos subpiales en las profundidades de los surcos, un patrón distintivo de la ETC.
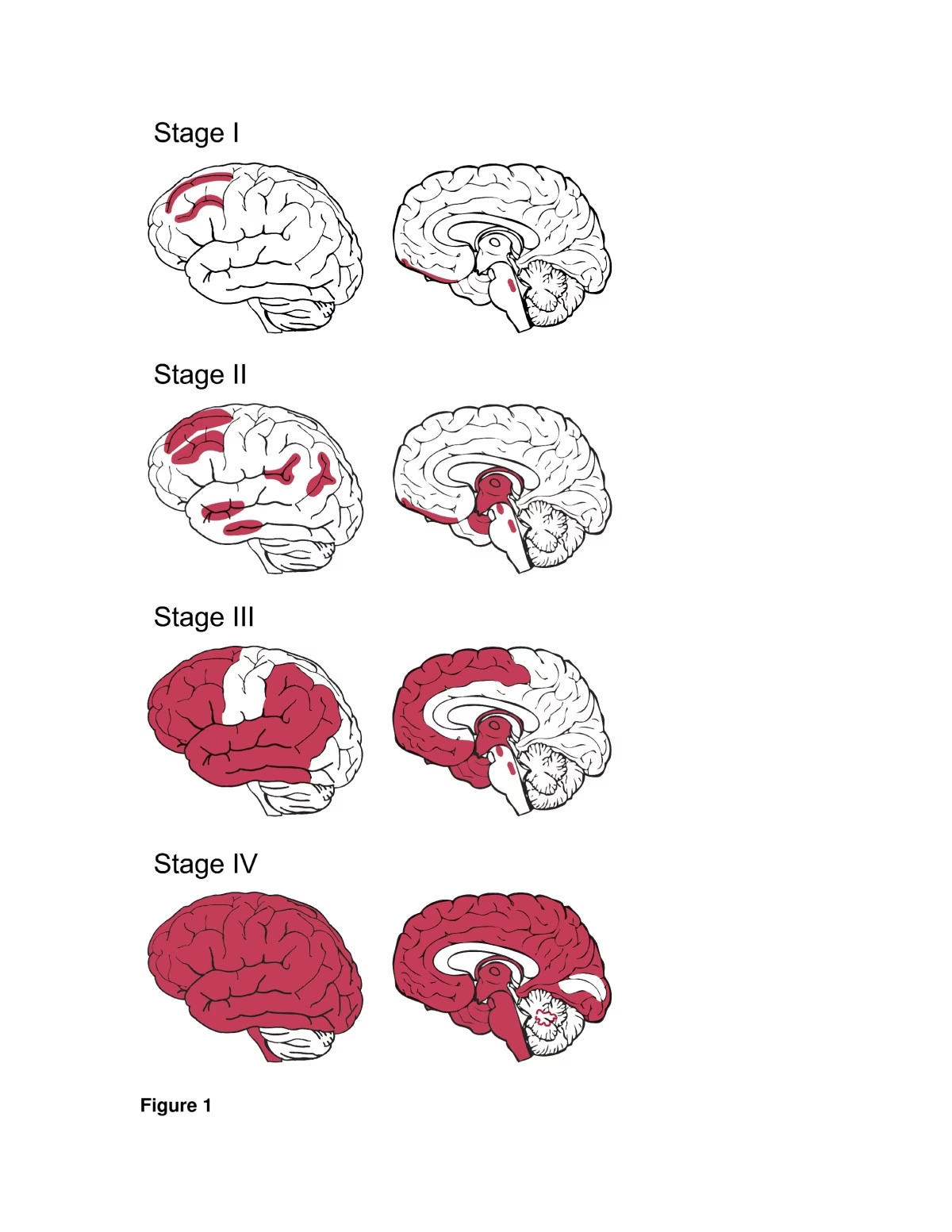
Estadificación de la Patología de Tau en la ETC
Utilizando los criterios provisionales, se ha propuesto un sistema de estadificación en cuatro etapas basado en la distribución y gravedad de la patología de p-tau. Este sistema, adaptado de la estadificación de Braak para la EA, correlaciona la gravedad patológica con factores como la duración de la carrera deportiva y la edad al fallecimiento.
A continuación, se presenta una tabla comparativa de las etapas de la ETC:
| Etapa de ETC | Hallazgos Macroscópicos Típicos | Patología de p-tau (Microscópica) | Patología de TDP-43 | Patología de Aβ |
|---|---|---|---|---|
| Etapa I | Generalmente normal; ocasional cavum septum pellucidum. | Focos perivasculares raros y aislados en profundidades de surcos (frontal, temporal, insular, septal, parietal); ONF y neuritas; ocasionales glía p-tau; ONF en locus coeruleus; OAs subpiales focales. | Raro, neuritas en sustancia blanca subcortical, fórnix (en ~50% de casos). | Ausente (a menos que >50 años). |
| Etapa II | Cambios sutiles: leve agrandamiento de cuernos frontales, 3er ventrículo; cavum septum pellucidum; palidez de locus coeruleus/sustancia negra. | Múltiples epicentros perivasculares en profundidades de surcos (frontal, temporal, parietal, insular, septal); ONF, pre-ovillos, neuritas, ocasional glía; OAs subpiales; ONF en capas corticales superficiales adyacentes; ONF en locus coeruleus, sustancia innominata. | Leve, neuritas o inclusiones aisladas en sustancia blanca subcortical, tronco encefálico, lóbulo temporal medial (subpial, periventricular, perivascular). | Presente en 19% (>50 años). |
| Etapa III | Reducción de peso cerebral; atrofia frontal/temporal leve; agrandamiento ventricular; anomalías septales (~50%); palidez de locus coeruleus/sustancia negra; atrofia de cuerpos mamilares, tálamo, hipotálamo; adelgazamiento cuerpo calloso. | Parches confluentes perivasculares en profundidades de surcos y capas superficiales; ONF, NTs, OAs; ONF en frontal, septal, insular, temporal, parietal inferior, bulbos olfatorios, hipocampo (CA4, CA2, CA1), corteza entorrinal, amígdala, hipotálamo, cuerpos mamilares, núcleo basal de Meynert, sustancia negra, núcleos del rafe, locus coeruleus; ocasional ONF en núcleo dentado cerebeloso, médula espinal (~33%). | Neuritas e inclusiones presentes en la mayoría de casos (corteza cerebral, lóbulo temporal medial, tronco encefálico). | Placas difusas y neuríticas dispersas en 13%. |
| Etapa IV | Disminución sustancial del peso cerebral (incluso <1000g); atrofia generalizada y pronunciada (frontal, temporal, lóbulo temporal medial, tálamo anterior); atrofia difusa de sustancia blanca; adelgazamiento cuerpo calloso; adelgazamiento hipotalámico severo; atrofia cuerpos mamilares; anomalías septales (cavum, perforaciones, ausencia posterior); palidez de locus coeruleus/sustancia negra. | Densamente distribuida (cerebro, tálamo, hipotálamo, cuerpos mamilares, ganglios basales, tronco encefálico, núcleo dentado cerebeloso, ocasional médula espinal); corteza visual primaria generalmente respetada; pérdida neuronal marcada (corteza, hipocampo, sustancia negra); microvacuolación en capa II cortical; ONF (muchos extracelulares) en formación hipocampal; patología de p-tau en cerebelo (núcleo dentado, capa granular, ocasional Purkinje); neuritas irregulares en sustancia blanca cerebelosa. | Widespread, en casi todos los casos; neuritas redondeadas/thread-like, inclusiones intragliales/intraneuronales (corteza cerebral, lóbulo temporal medial, diencéfalo, ganglios basales, tronco encefálico, ocasional médula espinal); acumulaciones densas en capa II cortical; inclusiones en fascia dentada (patrón DLFT-TDP). | No es un hallazgo universal. |
Patología de TDP-43 en la ETC
La patología anormal de la proteína TDP-43 es un hallazgo común y significativo en la ETC, presente en la mayoría de los casos, especialmente en las etapas más avanzadas. Consiste en la acumulación de TDP-43 mal plegada en el citoplasma de neuronas y células gliales, así como en neuritas distróficas. En las etapas iniciales, la TDP-43 se limita a neuritas en la sustancia blanca subcortical. Progresa a inclusiones aisladas en sustancia blanca, tronco encefálico o lóbulo temporal medial en la etapa II, a neuritas e inclusiones en corteza cerebral, lóbulo temporal medial o tronco encefálico en la etapa III, y se vuelve generalizada en la etapa IV, afectando múltiples regiones cerebrales y, ocasionalmente, la médula espinal.
La distribución de la patología de TDP-43 en etapas avanzadas, particularmente la afectación de la capa II cortical y la fascia dentada, se solapa con la observada en la DLFT-TDP. Esta superposición, junto con el desarrollo de enfermedad de motoneurona (EMN) en una proporción de individuos con ETC (aproximadamente 10%), sugiere que la ETC podría compartir mecanismos patogénicos con la DLFT-TDP.
Beta-Amiloide (Aβ) en la ETC
Los depósitos de beta-amiloide (Aβ), la proteína característica de las placas seniles en la enfermedad de Alzheimer, se encuentran en algunos casos de ETC, pero no son un hallazgo constante ni definitorio. La presencia de Aβ en la ETC está fuertemente asociada con la edad al fallecimiento. Aunque la deposición de Aβ parece ocurrir a una edad más temprana y a un ritmo acelerado en la ETC en comparación con el envejecimiento normal, y se asocia con una mayor gravedad clínica y patológica, la patología de Aβ no es necesaria para el diagnóstico de ETC.
Comorbilidades Neurodegenerativas
Al igual que otras enfermedades neurodegenerativas, la ETC a menudo coexiste con otras patologías neurológicas, especialmente en individuos mayores. Un porcentaje significativo de casos de ETC confirmados neuropatológicamente presentan patología adicional. Las comorbilidades más comunes incluyen enfermedad de motoneurona (EMN, incluyendo Esclerosis Lateral Amiotrófica - ELA), enfermedad de Alzheimer (EA), enfermedad por cuerpos de Lewy y degeneración lobar frontotemporal (DLFT). La coexistencia de estas patologías puede complicar el cuadro clínico y patológico, y determinar la contribución de cada una a los síntomas y la progresión es un desafío continuo.
La asociación entre ETC y EMN es particularmente notable, con aproximadamente el 10% de los casos de ETC desarrollando un síndrome de EMN clínicamente indistinguible de la ELA. Estos individuos a menudo presentan síntomas de EMN antes del deterioro cognitivo o conductual relacionado con la ETC y tienden a tener una patología de TDP-43 más severa que aquellos con ETC sola.
Relación entre Trauma Craneal Repetitivo y Patología
La fisiopatología exacta por la cual el trauma craneal repetitivo conduce a una neurodegeneración progresiva y taupatía latente en la ETC aún se está investigando. Se cree que las fuerzas de aceleración, desaceleración y rotación asociadas con los impactos o explosiones causan deformación y estiramiento de los elementos microestructurales del cerebro, como neuronas, células gliales y vasos sanguíneos. Estas fuerzas de cizallamiento afectan particularmente a las fibras largas (axones) y a los vasos sanguíneos, siendo más severas en puntos de cambio de dirección axonal o de densidad tisular (por ejemplo, alrededor de los vasos o en la interfaz sustancia gris-blanca).
Estudios experimentales y patológicos en humanos han demostrado que la lesión axonal y la disrupción vascular son más pronunciadas en puntos focales de estrés, como la región perivascular y las profundidades de los surcos corticales, precisamente donde se inicia la patología de p-tau en la ETC. La lesión microvascular y las microhemorragias, la astrogliosis y la activación microglial también se observan después de conmociones cerebrales agudas y subagudas, especialmente alrededor de pequeños vasos en la sustancia blanca.
El trauma experimental puede inducir la hiperfosforilación, mal plegamiento y agregación de la proteína tau. Sin embargo, cómo esta cascada de eventos (lesión axonal, disrupción de la barrera hematoencefálica, neuroinflamación, agregación de p-tau y TDP-43) conduce a una degeneración progresiva años o décadas después del último trauma sigue sin estar completamente claro. Es probable que factores adicionales como la susceptibilidad genética, la edad en el momento de la exposición, el género, el estrés, el abuso de sustancias y otras exposiciones ambientales jueguen un papel en la iniciación y progresión de la patología.
Propagación de la Patología de Tau
Aunque la fosforilación y el mal plegamiento de tau podrían ser procesos potencialmente reversibles en las etapas muy tempranas, las lesiones traumáticas repetidas y la acumulación de fragmentos tóxicos de p-tau parecen inducir un proceso auto-perpetuante. La neurodegeneración progresiva observada en atletas con ETC, que a menudo se manifiesta clínicamente años o décadas después de cesar la exposición al trauma, sugiere una propagación de la patología de tau a través del cerebro.
La evidencia experimental sugiere que la tau patológica puede transmitirse entre neuronas, posiblemente a través de un mecanismo similar a los priones, donde la tau mal plegada induce el mal plegamiento de la tau normal en las células receptoras. Esta propagación podría ocurrir a través de sinapsis neuronales o involucrar a células gliales (astrocitos, microglia) y vías de fluidos cerebrales como los espacios de Virchow-Robin perivasculares. La presencia de p-tau en estos espacios sugiere que los vasos sanguíneos no solo son un sitio de inicio de la patología, sino que también podrían ser una vía de propagación.
Patología Axonal
La lesión axonal, la degeneración axonal, la pérdida de fibras mielinizadas y la atrofia de la sustancia blanca son características constantes de la ETC. La lesión axonal probablemente desempeña un papel crítico en el inicio de la patología de p-tau. El grado de disrupción axonal se correlaciona con la gravedad de la neurodegeneración. En las etapas tempranas, se observan varicosidades axonales dispersas. En etapas más avanzadas (III y IV), la pérdida axonal es severa, con perfiles axonales distorsionados, acumulación de macrófagos y anomalías generalizadas de p-tau en la sustancia blanca.
Preguntas Frecuentes sobre la Neuropatología de la ETC
¿Qué causa la ETC a nivel cerebral?
La ETC es causada por el trauma craneal repetitivo, que desencadena la acumulación anormal de proteínas como la tau hiperfosforilada (p-tau) y TDP-43 en el cerebro. Estas proteínas se pliegan incorrectamente y se acumulan, dañando y matando las células cerebrales con el tiempo. La patología de p-tau se inicia de manera característica alrededor de los vasos sanguíneos en las profundidades de los surcos corticales.
¿Cómo se diferencia la ETC de la enfermedad de Alzheimer a nivel patológico?
Aunque ambas implican acumulación de proteína tau, la distribución es clave. En la ETC, la p-tau se acumula de forma característica en clusters perivasculares, especialmente en las profundidades de los surcos corticales y en las capas superficiales (II/III) de la corteza. En la EA, la patología de tau (ONF) comienza en el lóbulo temporal medial (corteza entorrinal, hipocampo) y se extiende de forma más predecible. Además, la patología de beta-amiloide es una característica definitoria de la EA, mientras que en la ETC está presente solo en algunos casos y se asocia a la edad, no siendo necesaria para el diagnóstico.
¿Se puede diagnosticar la ETC en vida?
Actualmente, el diagnóstico definitivo de la ETC solo se puede realizar mediante el examen neuropatológico del tejido cerebral post mortem. Aunque se están investigando biomarcadores e imágenes cerebrales para el diagnóstico en vida, aún no existen herramientas validadas para confirmar la ETC en personas vivas.
¿Qué papel juegan TDP-43 y Beta-Amiloide en la ETC?
La proteína TDP-43 se acumula de forma anormal en la mayoría de los casos de ETC, especialmente en etapas avanzadas. Contribuye a la patología y puede estar implicada en los síntomas, particularmente en aquellos que desarrollan enfermedad de motoneurona. La Beta-Amiloide se encuentra en aproximadamente la mitad de los casos, asociada a la edad, y puede agravar el cuadro, pero no es la patología definitoria de la ETC.
¿Cómo progresa la ETC a nivel cerebral?
La ETC progresa a través de etapas definidas por la distribución de la patología de p-tau. Comienza con focos perivasculares aislados en las profundidades de los surcos (Etapa I), se extiende a múltiples focos y capas superficiales (Etapa II), se vuelve más generalizada afectando el lóbulo temporal medial y otras regiones (Etapa III), y en la etapa más avanzada (Etapa IV), la patología de p-tau y TDP-43 es generalizada, causando atrofia cerebral severa y pérdida neuronal.
¿El daño cerebral por trauma puede curarse?
Aunque el cerebro tiene cierta capacidad de adaptación y plasticidad, especialmente en personas jóvenes, las células cerebrales destruidas o dañadas severamente generalmente no se regeneran. La recuperación después de una lesión cerebral traumática (LCT) puede implicar que otras áreas del cerebro compensen las funciones perdidas o que el cerebro aprenda a redirigir información. Sin embargo, la neurodegeneración progresiva de la ETC implica un daño continuo y no reversible a largo plazo.
En resumen, la neuropatología de la Encefalopatía Traumática Crónica es compleja y se caracteriza por la acumulación distintiva de p-tau en una distribución perivascular y en las profundidades de los surcos corticales, a menudo acompañada de patología de TDP-43. Estos hallazgos microscópicos, junto con los cambios macroscópicos observados en etapas avanzadas, reflejan el daño cerebral acumulado por el trauma craneal repetitivo y subyacen a la progresión clínica de la enfermedad. La investigación continua es fundamental para comprender completamente los mecanismos patogénicos, identificar la enfermedad en vida y desarrollar terapias efectivas.
Si quieres conocer otros artículos parecidos a La Neuropatología de la Encefalopatía Traumática Crónica puedes visitar la categoría Neurociencia.